")
")
| Issue |
Med Sci (Paris)
Volume 19, Number 2, Février 2003
|
|
|---|---|---|
| Page(s) | 187 - 199 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2003192187 | |
| Published online | 15 February 2003 | |
Des oncogènes aux régulateurs de la mitose : un changement de perspective dans notre vision des processus cancéreux
From oncogenes to mitotic regulators: a profound change in our view of the process of cancerous transformation
Institut de Génétique Moléculaire, Cnrs UMR 5535, IFR24, Équipe labellisée La Ligue, 1919, route de Mende, 34293 Montpellier Cedex 5, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
La vision que nous avons de la cellule tumorale a considérablement changé depuis la découverte des proto-oncogènes, gènes clés dont le dérèglement faisait du cancer une maladie de la signalisation cellulaire. En effet, l’émergence d’une cellule cancéreuse a progressivement été perçue comme résultant d’une augmentation de la fréquence des mutations, mutations qui sont ensuite sélectionnées par l’apparition d’un avantage sélectif, conduisant ainsi au concept du « phénotype mutateur ». L’instabilité génomique, reconnue comme mécanisme essentiel dans la genèse et l’évolution des processus cancéreux, et dont le rôle avait été entrevu depuis longtemps par les cytogénéticiens, transformait ainsi le cancer en une maladie de la réparation des lésions génotoxiques. Par ailleurs, l’instabilité chromosomique qui est fréquemment observée conduit à un nombre anormal de chromosomes (aneuploïdie) qui, très souvent, ont une structure aberrante. Les coupables, identifiés dans le camp des régulateurs du cycle cellulaire, provoquèrent un nouveau changement dans notre perspective, impliquant ainsi les mécanismes de contrôle de la bonne coordination des deux phases essentielles que sont la phase S et la mitose.
Abstract
Our vision of the cancer cell has dramatically changed since the discovery of proto-oncogenes, whose deregulation was proposed to mimic normal growth signalling. This notion, linking cancer to cell signalling pathways, has progressively led the way to the concept of the mutator phenotype, in which genetic instability plays an essential role in the onset of cancer. This then transformed cancer into a DNA repair disease. However, as foreseen decades ago by cytogeneticists, point mutations are not sufficient to give a full picture of the whole process. As a result, aneuploidy, rather than gene mutation, has been proposed as the explanation for the complex changes observed in cancer cells. The culprits were found among genes involved in the control of the cell division cycle, and work aimed at understanding the regulation of S phase and mitosis have yielded new insights into our understanding of cancer.
© 2003 médecine/sciences - Inserm / SRMS
Les poissons ont de si jolies têtes Qu’on est obligé de les déplacer fréquemment À cause du ravage qu’ils font dans le cœur des méduses
Raymond Queneau
Dire du cancer qu’il résulte d’un processus se déroulant en plusieurs étapes est maintenant un lieu commun. En effet, l’étiologie de cette pathologie suggère que la progression vers un phénotype tumoral agressif passe par l’acquisition d’au moins six propriétés qui pourraient servir à dresser un portrait robot de la cellule cancéreuse [1]: indépendance vis-à-vis des signaux de prolifération, insensibilité aux signaux antiprolifératifs, abolition de l’apoptose, capacité proliférative illimitée, capacité de susciter l’angiogenèse et acquisition d’un pouvoir invasif. L’apparition de ces traits spécifiques résulte de l’existence de mécanismes cellulaires intrinsèques dont le dysfonctionnement serait à l’origine du cancer. Toutefois, notre appréciation de leur importance relative a considérablement changé au cours des deux dernières décennies. Cet article se présente ainsi comme une saga destinée à donner une vue globale de l’évolution de nos idées sur les mécanismes moléculaires et cellulaires impliqués dans la survenue de tumeurs, où les principales observations et les concepts qui en ont découlé sont regroupés, pour des raisons didactiques, en trois actes.
Acte I : des proto-oncogènes aux gènes suppresseurs de tumeurs
La conversion proto-oncogène en oncogène vue comme une perversion des voies de signalisation
Les deux dernières décennies ont conduit tout d’abord à une vision du processus cancéreux comme une maladie de la signalisation inter- aussi bien qu’intracellulaire. Ainsi, parmi les oncogènes découverts à ce jour, nombre d’entre eux interviennent dans l’une des grandes voies de signalisation. À ce titre, les premiers travaux sur les rétrovirus transformants, en montrant que ces derniers avaient transduit des gènes cellulaires, ont joué un rôle majeur dans l’établissement du paradigme que constitue la signalisation mitogène sous le contrôle des MAP-kinases (Figure 1). En effet, en révélant que ces virus contenaient des formes mutées de gènes cellulaires, ces travaux, tout en fournissant un scénario pour le mécanisme de transformation virale, établissaient le concept de la conversion proto-oncogène → oncogène si communément utilisé de nos jours. Il devenait clair que de nombreux traits caractéristiques de la cellule cancéreuse étaient dus à la présence de mutations dites « gain de fonction » dans des gènes critiques favorisant un état permissif pour la prolifération. Ainsi, telle mutation conduit à une autocrinie, c’est-à-dire à la production par la cellule cancéreuse des facteurs de croissance dont elle a besoin, ou telle autre à la suppression du domaine extracellulaire régulateur d’un récepteur, qui de ce fait se comporte comme s’il était stimulé en permanence. Dans les deux cas, la cellule devient indépendante d’une intervention externe. De même, une mutation dans une protéine telle que Ras, qui sert de commutateur bipolaire associé à un récepteur, va la bloquer dans sa conformation active, avec pour conséquence une stimulation constitutive de la voie de signalisation efférente. Enfin, tel facteur de transcription, normalement présent en quantités infimes, à des moments précis ou dans des territoires particuliers, sera exprimé à des niveaux plus élevés, de façon anachronique ou encore ectopique, ce qui conduira à un changement quantitatif et qualitatif du programme génétique qu’il contrôle.
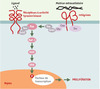 |
Figure 1. Le paradigme de la signalisation via les MAP-kinases et de sa coopération avec les intégrines. La fixation d’un ligand sur un récepteur doué d’activité tyrosine kinase conduit à la multimérisation de ce dernier et à son activation par autophosphorylation. Les tyrosines ainsi phophorylées au sein d’une courte séquence d’acides aminés constituent autant de cibles pour des protéines possédant des domaines susceptibles de les reconnaître, les domaines SH2 (Src homology domain 2) étant les plus représentatifs [55]. La fixation du ligand crée ainsi une surface d’interaction conditionnelle à la phosphorylation qui va alors recruter directement un effecteur ou passer par l’intermédiaire d’une molécule adaptatrice. L’activation de Ras est une parfaite illustration de ce dernier cas: il est ancré dans la membrane plasmique via un groupement farnésyl carboxy-terminal, et se trouve recruté par le récepteur via le complexe formé entre Grb2 et Sos. Grb2 est constitué d’un domaine SH3, associé à Sos de façon constitutive, et de deux domaines SH2, qui fixent des tyrosines phosphorylées dans des contextes appropriés. Ce faisant, Ras est mis en présence du facteur d’échange qu’est Sos, et passe de la forme se liant au GDP à celle se liant au GTP, active et capable d’interagir avec de nombreux effecteurs, dont Raf-1. Celui-ci constitue le premier terme d’une cascade de kinases constituée de trois modules, MAPKKK (Raf-1), MAPKK (MEK) et MAPK (ERK), cette dernière pouvant passer dans le noyau et phosphoryler des facteurs de transcription spécifiques. L’interaction de certaines intégrines avec la matrice extracellulaire peut également recruter les mêmes complexes et participer ainsi à l’activation de Ras. Les intégrines ne possédant pas d’activité kinase, plusieurs mécanismes peuvent être envisagés. Un mécanisme complexe, impliquant les kinases FAK (focal adhesion kinase) et src dans un jeu réciproque de phosphorylations, qui vont attirer une protéine adaptatrice telle que Grb2, est vraisemblablement impliqué dans la motilité cellulaire. Toutefois, la signalisation mitogène passerait par le recrutement de l’adaptateur Shc via la protéine membranaire cavéoline (Cav), la phosphorylation s’effectuant par l’un des membres de la famille Src. Quelques exemples parmi les premiers oncogènes viraux isolés: des facteurs de croissance comme sis (chaîne B du PDGF) ou int-2 (un variant du FGF); des kinases membranaires comme src, yes, lck, ou erbB (version tronquée du récepteur de l’EGF); des protéines G comme H-ras ou K-ras; des kinases cytoplasmiques comme raf/mil, mos ou pim-1; des facteurs de transcription comme myc, jun, fos ou erbA (récepteur de l’hormone thyroïdienne T3). |
Très vite, la prise en compte de la nécessité d’une coopération entre plusieurs voies de signalisation dans le maintien de l’homéostasie s’est imposée. Par exemple, la régulation de la prolifération et de la différenciation cellulaires fait intervenir à la fois des facteurs solubles, comme les facteurs de croissance, et l’adhérence à la matrice extracellulaire [2, 3] (Figure 1) ou à d’autres cellules au sein d’un même tissu (voir plus loin, Figure 6). Dans ce cadre, la cellule tumorale doit acquérir non seulement une autocrinie, mais également une indépendance vis-à-vis de la matrice extracellulaire, voire des cellules voisines. Par ailleurs, la cellule tumorale peut être amenée à « éduquer » son environnement par un dialogue avec les cellules avoisinantes, afin de favoriser certains processus tels que l’angiogenèse. Enfin, l’inactivation des mécanismes qui contrôlent l’apoptose va permettre à la cellule de s’affranchir des freins qu’imposent les systèmes de surveillance de son génome. Ainsi, la modification du fonctionnement de nombreuses voies de signalisation, dont certaines interviennent dans la différenciation cellulaire, peut concourir à l’établissement du phénotype tumoral. Ces deux derniers points sont en général avancés dans le cadre de ce modèle pour expliquer le caractère multiétapes des processus cancéreux, et la dédifférenciation fréquemment observée dans les tumeurs [1].
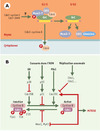 |
Figure 2. Contrôle des transitions G1/S et G2/M. A. Les origines de réplication (rectangles jaunes) fixent pendant la phase G1 le complexe hexamérique ORC (origin recognition complex), qui sert à son tour de plate-forme sur laquelle viennent se loger, de façon séquentielle, Cdc6, Cdt1, le complexe hexamérique MCM (mini-chromosome maintenance) (hélicase ?), et les facteurs requis pour le démarrage proprement dit de la réplication: Cdc45, RFA (replication factor A) et l’ADN polymérase α [56, 57]. L’assemblage et l’activation d’un tel complexe sont sous la dépendance, d’une façon qui n’est pas encore claire, des kinases Cdk2-cycline E et Cdc7-Dbf4. Les protéines Cdc6 et Cdt1 joueraient un rôle essentiel en recrutant ces facteurs afin de constituer le complexe de pré-initiation. Quand la réplication est enclenchée, Cdc6 et Cdt1 se détachent de la chromatine, à la suite d’une phosphorylation de Cdc6, vraisemblablement par Cdk2-cycline A. Cdc6 phosphorylé est exporté vers le cytoplasme. Quant à Cdt1, il est probablement inactivé par interaction avec la géminine. La reconstitution d’un complexe de préinitiation sur les origines qui ont été activées est ainsi évitée, et il ne peut, de ce fait, y avoir à nouveau réplication. Par ailleurs, sont également associées (mais non montrées sur ce schéma) des protéines de surveillance comme ATR (voir plus loin) qui vont arrêter la réplication en cas d’apparition de défauts au niveau de la fourche de réplication. B. Le complexe Cdk1/cycline B se forme à la suite de la synthèse de la cycline B en fin de phase S et de sa translocation depuis le cytoplasme (phases S et G2) vers le noyau (début de mitose). Son activité est contrôlée par une série de phosphorylations antagonistes. À la phosphorylation activatrice par la CAK (Cdk activating kinase) sur la thréonine 161 (boucle T, module l’interaction avec la cycline), s’opposent les phosphorylations inhibitrices sur la thréonine 14 et la tyrosine 15 (site de liaison de l’ATP) par les kinases Wee1 et Myt1 ([46] et Y. Pommier et K.W. Kohn, p.173 de ce numéro). La mutation de ces deux derniers acides aminés produit une entrée prématurée en mitose. Les résidus sérine 14 et tyrosine 15 sont déphosphorylés par les phosphatases Cdc25B et Cdc25C. L’activité et la localisation de ces phosphatases seraient également soumises à régulation, après phosphorylation par les kinases p38 pour Cdc25B, et Chk1 et Chk2 pour Cdc25C. Cette dernière, nucléaire juste avant la mitose, serait maintenue dans le cytoplasme à l’interphase, en raison de son interaction avec une protéine 14-3-3, après sa phosphorylation sur la sérine 216 par Chk1 et Chk2. Cdc25B contient plusieurs sites de fixation des protéines 14-3-3, dont la sérine 309, qui serait ciblée par la kinase de stress p38 en présence de lésions induites par les rayonnements ultraviolets [58]. Plk1 (Polo-like kinase) interviendrait également en amont et participerait à l’activation de Cdc25c. Quant aux autres phosphatases, elles doivent pour l’essentiel être caractérisées, PP2A supprimant vraisemblablement les phosphorylations apportées par les Cdk. La présence de lésions dans l’ADN active également une autre voie qui aboutit à l’inhibition de l’activité du complexe Cdk1-cycline B: l’induction de l’inhibiteur p21 par p53. |
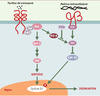 |
Figure 3. La production de la cycline D1, depuis sa synthèse jusqu’à sa dégradation, est sous le contrôle de stimulus externes. L’expression de la cycline D1 aurait pour finalité de faire sortir les cellules de leur état de quiescence et de rendre le cycle cellulaire dépendant de l’exposition des cellules à des facteurs de croissance, l’induction de la cycline E étant l’enjeu de ce processus. Ce dernier point est illustré par les expériences d’invalidation des gènes codant pour la cycline D1 ou la kinase Cdk4. Les souris déficientes en cycline D1 ou en Cdk4 viennent à terme, mais avec des problèmes de développement tels qu’une déficience oculaire, une taille réduite et une absence de maturation lobulo-alvéolaire terminale des glandes mammaires [7, 8]. Les animaux cycline D1−/−, chez lesquels la cycline E est réexprimée après insertion de son gène dans le locus cycline D1 (knock-in), ont un phénotype quasi normal. L’expression du gène cycline E est alors directement sous la dépendance des signaux extrinsèques, et ne nécessite plus une activation préalable de la cycline D1. Dans le même ordre d’idées, l’invalidation, dans les fonds génétiques cycline D1−/− ou Cdk4−/−, du gène codant pour p27Kip1, inhibiteur du complexe Cdk2-cycline E, supprime partiellement ces désordres [9]. Ce dernier résultat illustre le double rôle que joue le complexe Cdk4-cycline D1: activer la transcription du gène cycline E, et limiter la quantité libre de l’inhibiteur p27Kip1, avec pour résultat net une augmentation de l’activité Cdk2-cycline E. L’accumulation de la cycline D1 résulte, d’une part, de l’induction transcriptionnelle de son gène, et, d’autre part, de l’activation de la traduction de son ARN messager. Ces deux événements sont sous le contrôle de Ras via deux voies différentes: Raf/Mek/Erk et PI3-K/Akt/GSK-3β. Cette dernière voie contrôlerait également la localisation subcellulaire et la dégradation de la cycline D1. GSK-3β phosphoryle la thréonine 286 de la cycline qui est alors redirigée, pendant la phase S, vers le cytoplasme où elle est ubiquitinylée et dégradée par le protéasome. Akt, homologue cellulaire de l’oncogène v-Akt présent dans le virus AKT8, appartient à une famille de protéine kinases activées par les phospholipides engendrés par la PI-3K [59]. Elle voit donc son activité contrôlée par le suppresseur de tumeur PTEN (phosphatase and tensin homolog deleted on chromosome ten), capable d’hydrolyser les phospho-inositides. Elle est également phosphorylée par l’ILK (integrin linked kinase), liant ainsi la dégradation de la cycline D1 à l’adhérence cellulaire. |
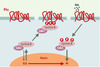 |
Figure 4. Interaction entre le suppresseur de tumeur Ptc et la cycline B1. La nævomatose basocellulaire, ou syndrome de Gorlin, est une maladie autosomique dominante prédisposant à l’apparition de nombreux cancers et anomalies du développement. Le gène de susceptibilité à ce syndrome a été localisé en 9q22.3, et la présence de déséquilibres alléliques sur ce site, observés dans les carcinomes basocellulaires aussi bien familiaux que sporadiques, en fait un gène suppresseur de tumeurs dont le mode de fonctionnement est encore inconnu. Des résultats récents montrent que Ptc peut interagir avec la cycline B1 [10]. Celle-ci est localisée dans le cytoplasme pendant les phases S et G2, puis dans le noyau en début de mitose, vraisemblablement grâce à une interaction avec la cycline F. Une séquence d’acides aminés en position amino-terminale, initialement caractérisée comme séquence de rétention cytoplasmique, fait office de séquence d’export nucléaire (SEN) qui, sous sa forme non phosphorylée (sérines 94, 96, 101, 113), permet son retour dans le cytoplasme par l’intermédiaire de CRM1 (chromosomal maintenance region 1). Plusieurs kinases sont susceptibles de phosphoryler le SEN: Cdk1-cycline B (Ser 94 et Ser 96) et Plk1 (Ser 113). La forme phosphorylée de la cycline B1 interagit avec Ptc en l’absence de son ligand, Shh, l’ajout de ce dernier provoquant la rupture de cette association. Ptc interagit aussi avec Smo (Smoothed) - une autre protéine trans-membranaire - qui serait en fait le composant chargé de la signalisation en aval. Cette observation ouvre ainsi deux possibilités non exclusives. D’une part, la rétention de la cycline B1 par Ptc dans le cytoplasme permet de retarder la mitose en l’absence de Shh, ce qui expliquerait le caractère suppresseur de tumeurs de Ptc. D’autre part, comme Smo est phosphorylé après liaison de Shh sur Ptc, qui n’est pas une kinase, le complexe Cdk1-cycline B1 pourrait être responsable de cette phosphorylation (cela n’est toutefois encore que pure spéculation). |
Ce schéma s’est enfin complété quand le caractère récessif de certains phénotypes tumoraux a été découvert. En effet, la possibilité de corriger certains de ces phénotypes par la réintroduction dans les cellules cancéreuses de fragments chromosomiques particuliers issus de cellules saines a montré l’existence de gènes dont la perte de fonction avait un caractère délétère. L’analyse des cohortes de cancers familiaux est venue conforter ces expériences conduites à partir d’hybrides somatiques. Le rétinoblastome, la tumeur de Wilms ou les polyposes adénomateuses familiales ont à ce titre constitué autant d’exemples mettant en exergue l’existence de gènes exerçant un effet négatif sur la prolifération. Ces gènes suppresseurs de tumeurs ont très vite été décrits comme appartenant eux-mêmes à des réseaux de signalisation intra- et intercellulaires, qui sont intimement imbriqués dans ceux impliquant les proto-oncogènes.
Une nouvelle connexion : les virus à ADN séquestrent ou détournent des régulateurs du cycle cellulaire
Un tel scénario de récupération d’un gène cellulaire n’existait pas dans le cadre de la transformation induite par les virus oncogènes à ADN: aucune séquence de leur génome ne semblait provenir de la cellule infectée, et leurs protéines régulatrices n’avaient pas de contrepartie évidente chez leurs hôtes. Pourtant, ces protéines étaient tout autant capables d’immortaliser des fibroblastes primaires et de coopérer entre elles pour transformer une cellule [4]. Toutefois, des protéines telles que E1A et E1B de l’adénovirus, T du SV40 ou du virus des polyomes (Py), et E6 et E7 des virus de papillomes étaient connues pour s’associer à des protéines cellulaires. Alors que le génome du Py code pour trois protéines régulatrices (t, τ et T), seulement deux protéines (t et T) étaient connues chez le SV40, pourtant très proche du Py. La recherche chez SV40 d’une protéine cousine de τ a conduit à la caractérisation de ce qui devait devenir le fameux suppresseur de tumeur, p53. Émerge alors un autre scénario: au lieu d’introduire dans la cellule infectée une version déréglée, constitutivement active, d’une protéine qui va lui conférer un caractère permissif pour la prolifération, les virus à ADN séquestrent des protéines cellulaires qui, pour la plupart, ont un rôle antiprolifératif. Autrement dit, on retrouve les gènes suppresseurs de tumeurs mentionnés précédemment. De plus, l’examen des protéines piégées par leur association avec les protéines virales révèle que ce sont des régulateurs du cycle de division cellulaire: p53, mais également pRb, le produit du gène de susceptibilité au rétinoblastome, ou la cycline K, dans le cas du virus herpétique HHV8.
Brièvement, le cycle cellulaire se caractérise par l’articulation coordonnée de deux événements majeurs pour la cellule: la duplication de son ADN, ou phase S, et la ségrégation correcte des deux lots de chromosomes qui en résultent, ou mitose (ou encore phase M). Ces deux phases sont elles-mêmes séparées par deux phases, G1 et G2, dont la durée varie durant l’ontogenèse, et suivant les organismes [5]. Les diverses transitions entre les phases sont sous le contrôle d’une famille de protéine kinases constituées de trois sous-unités: une sous-unité catalytique (Cdk: cyclin-dependent kinase), une sous-unité activatrice (la cycline) et une sous-unité inhibitrice (les inhibiteurs de Cdk [CKI] tels que p15, p16, p21 ou p27), toutes trois appartenant à des familles protéiques qui ne sont pas toutes impliquées dans le contrôle du cycle cellulaire [6] Le cycle cellulaire peut arbitrairement être divisé en deux modules. Le premier, dévolu au contrôle des deux phases essentielles que sont S et M, correspond à un mécanisme intrinsèque sous le contrôle des complexes Cdk2-cycline E/A et Cdk1-cycline A/B (Figure 2). Le second module permet de coupler le cycle cellulaire à des facteurs extrinsèques responsables de l’homéostasie tissulaire. Ainsi, au cours de la transition de l’état de repos vers la prolifération, et par l’intermédiaire des kinases Cdk4 et Cdk6 associées aux cyclines D, le cycle cellulaire peut être modulé par l’exposition des cellules à certaines cytokines ou autres facteurs issus des cellules voisines, ou même de la matrice extracellulaire (Figure 3). Des facteurs extrinsèques peuvent également, semble-t-il, suspendre la progression du cycle cellulaire pendant la transition G2/M. Ainsi, des résultats récents suggèrent que la cycline B1 peut interagir avec le suppresseur de tumeur Patched 1 (Ptc1) [10]. Ce dernier est le récepteur transmembranaire de la protéine Sonic hedgehog (Shh), morphogène spécifiant l’axe dorso-ventral du tube neural, ainsi que l’axe antéro-postérieur des ébauches des membres et du tube digestif chez les vertébrés. Le gène Ptc1 est fréquemment inactivé dans les nævomatoses basocellulaires, prédisposition familiale à certains cancers de la peau (syndrome de Gorlin), tandis que Shh est un oncogène puissant dans les modèles de souris transgéniques. La forme phosphorylée de la cycline B1 interagirait avec Ptc1, qui la séquestrerait ainsi dans le cytoplasme (Figure 4). À l’opposé, la fixation de Shh sur son récepteur libérerait la cycline B1 de cette interaction. Or, il faut savoir que le domaine d’interaction de la cycline B1 avec Ptc1 est impliqué dans la localisation subcellulaire de la cycline, qu’il contrôle précisément par son état de phosphorylation. Bien que la signification de cette interaction ne soit pas encore comprise, cette observation ouvre plusieurs perspectives très intéressantes sur le rôle suppresseur de tumeur de Ptc1.
Ces différents protagonistes sont retrouvés à divers titres dans de nombreuses néoplasies. L’inactivation des gènes codant pour p16 et p15 est très fréquente dans les lignées cellulaires cancéreuses et les tumeurs dont elles sont issues [11]. En outre, les souris chez lesquelles le locus INK4A, codant pour p15 et p16, entre autres protéines, a été invalidé, développent très rapidement de nombreuses tumeurs et sont très sensibles aux agents carcinogènes. Les protéines p16 et p15 sont des inhibiteurs des complexes kinases Cdk4, Cdk6-cycline D, dont la cible privilégiée est la protéine Rb, elle-même souvent mutée dans les cancers humains. L’action de p15 et de p16 requiert une protéine Rb fonctionnelle, et toute inactivation de cette dernière rend ces deux inhibiteurs inopérants. Par ailleurs, l’expression de p15 est sous le contrôle du TGF β (transforming growth factor β) , ce qui fait du locus INK4A-INK4B une plaque tournante des actions du milieu extérieur sur le cycle cellulaire, via la protéine Rb.
Les cyclines E et D1 sont présentes à des niveaux élevés dans de nombreuses lignées cancéreuses, et le gène humain de la cycline D1 (c’est-à-dire PRAD1) a été isolé à la suite de la caractérisation de remaniements chromosomiques survenant dans certaines tumeurs du sein [12]. En outre, les souris transgéniques exprimant la cycline D1 seule [13], ou en conjonction avec c-Myc [14, 15], de façon ciblée vers la glande mammaire ou le tissu lymphoïde, développent respectivement des adénocarcinomes et des lymphomes. À l’opposé, les souris dont le gène de la cycline D1 a été invalidé (D1−/−, voir légende de la Figure 3) sont devenues résistantes à l’expression forcée, dans les glandes mammaires, de certains oncogènes tels que HER-2/erbB-2/neu ou ras, connus pour provoquer l’apparition de tumeurs de ce tissu chez la souris [16].
Acte II : oui, mais alors il faut des mutations! Du phénotype mutateur à l’instabilité chromosomique
La conversion proto-oncogène → oncogène est soustendue par l’existence de mutations résultant soit d’une réparation défectueuse des lésions qui se produisent naturellement dans l’ADN, soit d’une mauvaise détection de ces dernières. Or, le bon déroulement du cycle de division cellulaire nécessite la mise en place d’une série de contrôles de qualité (checkpoints). De nombreux travaux ont confirmé le rôle très important joué par les dysfonctionnements des mécanismes contrôlant la stabilité et l’intégrité génomiques.
Réparation défectueuse : mutations dans les gènes MSH et MLH
Si l’on prend le cas des cancers coliques non polyposiques, on trouve à l’origine de 50 % d’entre eux une fréquence élevée de mutations ponctuelles ou de microdélétions dues à une réparation défectueuse des mauvais appariements de paires de bases dans l’ADN. Cela se traduit par une instabilité au niveau des microsatellites (MIN) (→), phénomène d’ailleurs utilisé dans le diagnostic de cette maladie. Ces désordres résultent de mutations apparaissant dans la lignée germinale et touchant les gènes hMSH et hMLH (human MutS et MutL homologs), qui sont les homologues humains des gènes bactériens MutS et MutL [17, 18] (→→). Les protéines codées par ces derniers forment des complexes macromoléculaires ayant pour fonction de reconnaître des appariements incorrects dans l’ADN. Ces derniers apparaissent au cours de la réplication ou de la recombinaison, ou encore lors d’une exposition à des agents génotoxiques. Ils résultent soit de l’ajout ou de l’élimination d’une base, soit de l’incorporation d’une base incorrecte, soit encore d’une altération chimique comme la désamination de la 5-méthyl cytosine, par exemple. La recherche de gènes de susceptibilité a conduit à l’identification de certains d’entre eux sur le chromosome 2. De fait, la réintroduction de ce dernier dans des cellules humaines mutées sur les gènes hMSH2 et hMSH6 abroge leur hypermutabilité et restaure leur sensibilité à la nitrosoguanidine, un agent mutagène [19, 20]. L’effet de l’invalidation de ces gènes chez la souris a conforté leur rôle dans l’hypermutabilité observée dans les tumeurs humaines, et de fait leur participation à l’oncogenèse [21–23].
(→) m/s 2002, n°11, p. 1058
(→→) m/s 2003, n°1, p. 85
Détection défectueuse des lésions de l’ADN : mutations des membres de BASC
Par ailleurs, les protéines évoquées ci-dessus ont été détectées dans le complexe supra-moléculaire BASC (BRCA1-associated genome surveillance complex) [24], qui contient également d’autres suppresseurs de tumeurs, parmi lesquels BRCA1 (breast cancer 1) ou ATM (ataxiatelangectasia mutated), intervenant dans la détection de lésions sur l’ADN (Figure 5). Des mutations germinales dans les gènes codant pour BRCA1 et BRCA2 rendent compte de 50 % des cancers du sein familiaux [25–29]. En outre, des mutations de BRCA1 sont retrouvées dans la quasi-totalité des syndromes familiaux associant cancers du sein et de l’ovaire. Signalons enfin la découverte récente que le gène de prédisposition à l’anémie de Fanconi, FANCD1 est identique au gène BRCA2 [60] (→). Quant au gène ATM, il prédispose à l’ataxie-télangiectasie, maladie se caractérisant, entre autres, par une hypersensibilité aux radiations ionisantes et un risque élevé de cancers [30,31]. La protéine ATM appartient à une famille de kinases présentant des similitudes de structure, comme ATR (ATM-related) ou DNA-PK (DNA-dependent protein kinase), qui jouent un rôle de relais essentiel entre les protéines détectant les lésions et les effecteurs de la réparation et du cycle cellulaire. En cas de lésions induites en phase G1 par les rayons ultraviolets ou d’autres agents clastogènes, ATM et ATR « se disputent les faveurs » de p53 en phosphorylant directement, ou indirectement via Chk2, son extrémité amino-terminale. Cela a pour double conséquence de stabiliser p53 et d’augmenter sa capacité transactivatrice [31]. En phase G2, c’est Cdc25C, la phosphatase qui prépare le complexe Cdk1-cycline B à la mitose, qui devient une cible des kinases ATM et ATR, via Chk1 et Chk2. Pour résumer, BASC constituerait une unité multifonctionnelle de surveillance de l’intégrité du génome, constituée de plusieurs modules. Un module de détection des lésions serait ainsi associé, via MSH2/MSH6, à un module de signalisation qui fonctionnerait comme un commutateur moléculaire utilisant l’hydrolyse de l’ATP, à la manière dont Ras utilise le GTP [32–34].
Ces diverses observations ont conduit au concept de « phénotype mutateur », qui voit l’émergence d’une cellule cancéreuse comme la résultante d’une augmentation de la fréquence des mutations, ensuite sélectionnées par l’apparition d’un avantage sélectif (autrement dit, d’un avantage conféré à une cellule mutée par rapport aux cellules normales, dans les conditions particulières qui règnent au sein de la tumeur: par exemple, la capacité de proliférer en milieu fortement anoxique). L’instabilité génomique, reconnue comme mécanisme essentiel dans la genèse et l’évolution du processus cancéreux, un rôle entrevu depuis longtemps par les cytogénéticiens, transforme ainsi celui-ci en une maladie de la réparation des lésions génotoxiques.
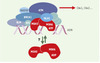 |
Figure 5. BASC, un complexe de surveillance de l’intégrité de l’ADN ? BASC (BRCA1- associated genome surveillance complex) [24] est constitué par une quarantaine de protéines déjà connues pour s’associer à BRCA1, telles BARD1 (BRCA1-associated ring domain) ou HDAC1 (histone deacetylase 1), ainsi que par des protéines impliquées dans la détection de lésions, comme le complexe MSH2-MSH6 ou encore ATM, et dans la recombinaison homologue, comme le complexe RAD50-MRE11-NBS1. Sont également présentes les trois sous-unités du facteur de réplication RFC impliqué dans la mise en place de PCNA (proliferating cell nuclear antigen, un co-facteur de l’ADN polymérase δ) sur la fourche de réplication. Pour le moment, on ne sait pas si BASC est constitué d’un seul complexe, ou de plusieurs sous-complexes dévolus à des tâches différentes. De même, le rôle de MSH2-MSH6 est ouvert: soit il est impliqué dans le recrutement de BASC après avoir reconnu la lésion, soit il est toujours associé à BASC, et c’est un changement de conformation, lié à l’hydrolyse de l’ATP, qui joue un rôle dans la signalisation (ces deux hypothèses n’étant pas exclusives) [24–30]. Les gènes MSH ont été invalidés chez la souris et les cellules issues de ces animaux montrent une instabilité des satellites, une tolérance vis-à-vis des agents alkylants et, de façon intéressante, un laxisme de la recombinaison homologue visà- vis de l’identité des séquences d’ADN impliquées. Les animaux MSH2−/− ne présentent pas d’anomalies majeures, mais 30 % d’entre eux développent des lymphomes à un âge précoce [21]. De même, les souris MSH6−/− développent toutes des lymphomes et des tumeurs cutanées ou utérines, cette distribution pouvant changer avec le fond génétique des animaux utilisés, suggérant ainsi que les complexes MSH2/MSH6 et MSH2/MSH3 ont des spécificités différentes in vivo. |
Acte III : vous avez dit APC ou APC ? Histoire d’une homonymie troublante qui nous mène vers une vision plus cellulaire
Cependant, par rapport au phénotype Min (voir plus haut), une instabilité au niveau chromosomique (CIN) est observée dans une grande majorité de cancers, tous types de cancers confondus, qui conduit à un nombre anormal de chromosomes (aneuploïdie) ayant très souvent une structure aberrante. Les responsables sont, dans ce cas, à rechercher dans le camp des régulateurs du cycle cellulaire ou de ses constituants, qu’ils contrôlent la transition G1/S ou la mitose. Ainsi, une expression forcée du complexe Cdk2-cycline E dans les cellules en culture conduit à une instabilité chromosomique. Il en est de même (voir plus loin) pour plusieurs régulateurs de la mitose. Nous retrouvons donc là une implication des mécanismes qui contrôlent la bonne coordination des deux phases essentielles que sont la phase S et la mitose. Les travaux récents réalisés sur cette dernière conduisent à un nouveau changement dans la vision de la cellule cancéreuse. En effet, les processus étaient jusqu’à présent envisagés pour eux-mêmes, indépendamment de la structure qui les supportait. Les données actuelles prennent maintenant en compte la structure cellulaire, et font en particulier jouer un rôle essentiel au cytosquelette et au fuseau mitotique, dont la structure et la dynamique sont des paramètres extrêmement importants.
Dynamique du cytosquelette et cancer
Comme nous l’avons vu plus haut, l’analyse génomique des tumeurs coliques de petite taille montre que le déséquilibre allélique y est très fréquent: plus de 90 % des tumeurs analysées présentent un tel déséquilibre sur au moins un chromosome, et 67 % sur un chromosome autre que le 5 [35]. Rappelons que ce dernier abrite en 5q21 le gène APC (adenomatous polyposis coli), muté lui-même dans 80 % des cas de polypose adénomateuse familiale et dans plus de 60 % des cancers sporadiques du côlon. La protéine APC est connue pour son rôle de modulateur de la voie de signalisation, sous le contrôle de Wnt-1 [36–38].
En l’absence de signal émanant de Frizzled, récepteur de Wnt-1, APC forme un complexe contenant la caténine β. Cette dernière est alors dégradée par le protéasome après ubiquitinylation consécutive à sa phosphorylation par GSK-3β. En présence de Wnt-1, la caténine β libre n’est plus phosphorylée et, n’étant plus dégradée, peut migrer dans le noyau où elle forme, avec Tcf (T-cell factor, ou encore lymphoid-enhancer factor: LEF), un complexe transcriptionnel actif (Figure 6), dont les gènes cibles potentiels sont cycline D1 et le proto-oncogène c-myc. APC contrôle l’activité de ce complexe à deux niveaux, celui de la translocation nucléo-cytoplasmique de la caténine et celui de sa dégradation. Les mutations observées sur APC suppriment ces deux fonctions et, en permettant à la caténine β de s’accumuler dans le noyau (la caténine n’est plus dégradée en dehors des jonctions adhérentes, et sa translocation n’est plus modulée et devient constitutive), miment ainsi une stimulation constitutive de la voie Wnt-1/Frizzled. La grande majorité des tumeurs coliques contient deux allèles APC inactivés, alors qu’une minorité présente des mutations activatrices sur la caténine β ou des troncatures de l’axine, une autre protéine du complexe APC/caténine β. Toutefois, les mutations sur la caténine β semblent plus fréquentes dans les adénomes de petite taille (12,5 %) que dans ceux de grande taille (24 %), et sont rares dans les carcinomes invasifs, suggérant ainsi que les mutations sur les protéines APC et la caténine β ne sont pas fonctionnellement équivalentes [39]. La caténine β est également présente au niveau des points de jonction entre cellules, en interaction avec le cytosquelette d’actine via la caténine α. APC, en contrôlant le destin intracellulaire de la caténine β, occupe ainsi une place centrale dans la régulation de la dynamique du cytosquelette.
Les données récentes [40, 41] suggèrent que ceci n’est qu’une partie de l’histoire: APC, qui est également trouvé à l’extrémité en croissance des microtubules [42, 43], pourrait aussi jouer un rôle dans le contrôle de l’attachement des microtubules aux chromosomes. L’interaction avec ces derniers est vraisemblablement complexe, et il ne faut pas perdre de vue qu’il existe au moins deux variants d’APC qui, s’ils fonctionnent tous deux dans la voie Wnt-1, fixent des protéines différentes. De façon remarquable, la plupart des mutations d’APC se trouvent dans sa partie carboxy-terminale, qui est impliquée dans l’interaction avec les kinétochores, structures responsables de l’attachement des chromosomes au fuseau mitotique (Figure 6). APC y forme un complexe avec les protéines Bub1 et Bub3, initialement découvertes au cours d’un criblage destiné à rechercher des mutants insensibles aux substances qui dépolymérisent les microtubules. Ces protéines (Bub1, Bub2, Bub3, BubR1/Mad3, Mad1, Mad2) assurent le déroulement correct de la mitose (spindle checkpoint) en contrôlant deux transitions essentielles: la transition métaphase-anaphase et la sortie de mitose [44, 45]. La phosphorylation d’APC, qui est en fait un très bon substrat pour les kinases du complexe Bub/Mad, diminue sa capacité de se lier aux microtubules. Les mutations dans APC se traduisent par une perte de son association avec les kinétochores, avec pour conséquence une ségrégation anormale des chromosomes. Toutefois, certaines de ces mutations n’affectent pas la capacité d’APC d’induire la dégradation de la caténine β, ce qui suggère que les effets observés ne sont pas transcriptionnels. Par ailleurs, une expression ectopique du seul domaine carboxy-terminal multiplie grandement les anomalies chromosomiques. En outre, les cellules APC−/− présentent des centrosomes surnuméraires. Ainsi, deux types de défauts s’accumulent dans ces cellules: quantitatifs, avec une polyploïdie due à une absence de disjonction des chromatides soeurs, et qualitatifs par l’apparition de réarrangements chromosomiques.
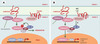 |
Figure 6. Voie de signalisation Wnt/Frizzled/caténine β. A.La caténine β est soit libre, soit engagée dans un complexe avec la caténine β et les cadhérines au niveau des jonctions adhérentes. En l’absence de Wnt, la caténine β libre est dégradée par le protéasome, via un complexe de destruction contenant APC, l’axine et la glycogène-synthétase-kinase-3 β(GSK-3 β). Cette dernière phosphoryle la caténine β, reconnue alors par le complexe SCF (Skip1- Culin-Fbox) via la protéine F-box β-TrcP, ce qui entraîne son ubiquitinylation et sa dégradation. Ce faisant, la transcription des gènes contrôlés par LEF/Tcf (lymphoid-enhancer factor/T-cell factor) est silencieuse, ce dernier étant inhibé dans le noyau par le co-répresseur Groucho. B. En présence de WNT, le récepteur Frizzled, associé à une protéine apparentée au récepteur des lipoprotéines de basse densité (LRP6), est activé. S’ensuit une cascade d’événements peu documentée au cours de laquelle l’inhibiteur de GSK-3β GBP est activé via Dishevelled (pour plus de détails, voir [38]). En conséquence, la caténine β n’est plus dégradée, diffuse dans le noyau, déplace Groucho et active la transcription sous le contrôle de LEF/Tcf. APC contrôle ainsi le destin intracellulaire de la caténine β en orchestrant sa distribution entre les jonctions adhérentes, le cytoplasme et le noyau. |
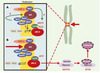 |
Figure 7. APC et APC/C se retrouvent dans les kinétochores. APC (adenomatous polyposis coli) et APC/C (anaphase promoting complex) se retrouvent au niveau des kinétochores, structures chargées du lien entre les chromosomes et les microtubules constituant le fuseau mitotique (flèches vertes épaisses). APC est soit impliqué dans le contrôle, soit est un acteur essentiel de la dynamique des microtubules. Il est phosphorylé, au moins in vitro, par les kinases Bub1-Bub3 et BubR1-Bub3. Celles-ci sont vraisemblablement également impliquées dans le recrutement de Mad2 par Mad1 ou APC/C, ce dernier étant inactif tant que tous les kinétochores ne sont pas engagés dans une liaison et que tous les chromosomes ne sont pas correctement alignés (A). Quand cela se produit, un signal encore mal identifié (structure de la jonction microtubule-kinétochore ? tension du fuseau sur le kinétochore ?) est interprété via CENP-E (centromeric protein E) en termes de phosphorylations par les kinases Bub1-Bub3, BubR1-Bub3 et Mps1. Mad2 se dissocie alors de APC/C, qui est ainsi activé (B). Cdc20/Fizzy/p55CDC est une protéine adaptatrice qui permet à APC/C de diriger la sécurine vers le protéasome après son ubiquitinylation, ce qui a pour effet d’activer la séparase, protéase responsable de la dégradation des cohésines. Ces dernières forment un complexe d’au moins quatre sous-unités, responsable de l’attachement entre les chromatides soeurs. Plusieurs autres kinases interviennent très vraisemblablement. Ainsi, lors d’une lésion dans l’ADN, Chk1 phosphoryle la sécurine, ce qui la rend insensible à APC/C. Mais l’on trouve également au niveau du kinétochore MAPK activée, Plk1 et Aurora B, qui semblent également intervenir dans la cytodiérèse, mais pour lesquelles il est encore difficile de définir un rôle exact. Les gènes Plk1, Aurora A, Aurora B et Aurora C sont associés à des locus chromosomiques altérés dans de nombreux cancers. Aurora A et Plk1 sont surexprimées dans certaines tumeurs primaires, et sont capables de transformer des cellules en culture. |
Mauvais fonctionnement du fuseau mitotique et processus cancéreux
De façon étonnante, l’acronyme APC désigne également un complexe multi-enzymatique (anaphase promoting complex) qui, chez les vertébrés, est localisé presque essentiellement sur le fuseau mitotique et dans les kinétochores (Figure 6). Dans la suite du texte, il sera désigné par l’acronyme APC/C, pour APC/cyclosome. APC/C est doté d’une activité ubiquitine ligase dévolue à la dégradation de protéines de régulation de la mitose, telles que les cyclines A et B et les sécurines (Pds1 chez Saccharomyces cerevisiae), protéines qui orchestrent la séparation des chromatides soeurs [48]. Brièvement, les chromatides, condensées pendant la pro-métaphase, établissent une liaison bipolaire avec le fuseau mitotique émanant des centrosomes, via leur kinétochore. Si l’un d’entre eux n’est pas attaché, un signal, dont l’origine n’est pas encore connue (tension du fuseau ?), est émis, qui va retarder la transition vers l’anaphase, permettant ainsi aux retardataires de rejoindre le lot sur la plaque équatoriale. Ce signal est intégré par le complexe Bub/Mad, associé au moteur moléculaire qu’est la kinésine CENP-E, lien mécanique entre microtubules et kinétochore. La cible de ce signal est APC/C, qui est maintenu dans une forme inactive jusqu’à ce que les derniers liens soient assurés. APC/C peut alors dégrader la sécurine, inhibiteur de la séparase, qui va elle-même dégrader les protéines chargées du maintien de la cohésion entre les chromatides soeurs, les cohésines.
Chez la levure, des mutations dans l’un des membres de ce module de contrôle du bon fonctionnement du fuseau mitotique se traduit par une perte rapide de chromosomes. Il est clair que ce module est affecté dans certains cancers qui ne répondent pas aux substances antagonistes du fuseau mitotique, et où l’on trouve des mutations dans les gènes Bub1 ou Mad2 [49, 50]. Il est de ce point de vue particulièrement intéressant d’observer que, dans une cellule compétente pour le point de contrôle mitotique, l’introduction d’un allèle muté de Bub1 entraîne l’inactivation de ce point de contrôle, montrant le caractère dominant de la mutation [49]. L’invalidation de Bub3 ou de Mad2 chez la souris conduit à une létalité très précoce, et des cellules Mad2+/- en culture montrent une instabilité chromosomique due à une entrée prématurée en anaphase [51, 52]. De la même façon, des cellules humaines dans lesquelles ont été invalidés, par recombinaison homologue, les deux allèles du gène hSécurine, présentent des mitoses anormales et une fréquence élevée de perte de chromosomes [53]. Or, hSécurine avait déjà été identifié comme un gène de prédisposition à des tumeurs de l’hypophyse (PTTG, pituitary tumour transforming gene), capable d’immortaliser des fibroblastes et de conduire à des tumeurs chez des animaux athymiques.
Conclusions
Nous avons vu que l’interprétation des mécanismes conduisant une cellule à engager un processus cancéreux a progressivement évolué. La découverte des versions virales (v-onc) de gènes cellulaires capturés par des rétrovirus a conduit à la notion de proto-oncogène. Ces derniers, à la suite de changements qualitatifs ou quantitatifs, sont exprimés à des niveaux très élevés, de façon anachronique ou encore ectopique, ce qui favorise une prolifération cellulaire mal contrôlée et excessive. Par ailleurs, la recherche par clonage positionnel de gènes de susceptibilité a contribué à enrichir cette vision, en révélant l’existence de gènes dont c’est l’absence ou l’inactivation qui confère cet avantage prolifératif. Proto-oncogènes et gènes suppresseurs de tumeurs ont ainsi constitué le yin et le yang du contrôle de la prolifération. Le premier cercle de ces gènes très spéciaux s’est très vite élargi, à tel point que le qualificatif d’oncogène ou de gène suppresseur devient parfois d’ordre purement sémantique. Par exemple, de nombreux ARN messagers cellulaires traduits de façon peu efficace codent pour des fonctions importantes dans la prolifération. Cette traduction peu efficace résulte en une faible accumulation des protéines. Il en ressort qu’une élévation anormale de la quantité d’eIF4E, en augmentant l’efficacité de la traduction, va conduire de façon indirecte à une surproduction de ces protéines, et donc à une prolifération déréglée. Ainsi, la simple expression forcée d’eIF4E, une protéine impliquée dans les premières étapes de la traduction, et qui est présente en quantités limitantes, peut conférer un caractère permissif pour la prolifération [54]. Ces difficultés sémantiques ont conduit certains auteurs [3] à classer les altérations génétiques présentes dans une cellule tumorale en regroupant les gènes impliqués en gatekeepers, caretakers et landscapers (dont la traduction littérale, qui n’est pas forcément très satisfaisante, serait « sentinelles ou garde-barrières », « gardiens » et « ajusteurs ou modulateurs de plasticité »). Il s’agit de dépasser la classification oncogènes/gènes suppresseurs de tumeurs, finalement limitée et très imparfaite.
Par ailleurs, la vision qualitative, présence ou absence d’un gène, correspondant à l’hypothèse de Knudsen qui stipule qu’un seul événement suffit à faire émerger un oncogène, alors que deux épisodes sont requis pour annihiler la fonction d’un gène suppresseur, est malmenée. En effet, de nombreux cas d’haplo-insuffisance ont été découverts, et il est maintenant évident que l’action de certains gènes est extrêmement sensible à la quantité des produits qu’ils engendrent, certaines mutations (de TGFβ ou p27, par exemple) se faisant déjà ressentir à l’état haploïde.
En outre, plusieurs expériences d’invalidation génique ont conduit à des résultats diamétralement opposés à ceux qui étaient attendus. Si la raison de cette déconvenue réside en partie dans le fait que les données obtenues in vitro ou ex vivo ne se transposent pas toujours in vivo, elle est très certainement à méditer dans le cadre du fonctionnement en réseaux multimodulaires des diverses voies de signalisation impliquées. En effet, ces dernières se caractérisent par l’existence de nombreuses boucles de régulation, caractéristiques des phénomènes complexes non linéaires. Ces boucles exercent une action en retour dont l’effet peut être négatif, avec très souvent pour résultat de limiter une réponse dans le temps, ou positif, avec pour conséquence une amplification transformant une réponse graduelle en un effet de « tout ou rien ». Ainsi, p53, en induisant Mdm2,conduit à sa propre destruction. À l’opposé, le facteur de transcription c-Jun, en se fixant sur le promoteur de son propre gène, contribue à amplifier sa production. On comprend alors que des réponses fort différentes soient engendrées suivant le niveau auquel est exprimé un effecteur critique.
Il restait à comprendre les mécanismes générateurs des mutations responsables, d’une part, de la conversion des proto-oncogènes en oncogènes actifs, et, d’autre part, de l’inactivation des gènes suppresseurs. De nombreuses observations avaient conduit les cytogénéticiens à proposer que les anomalies chromosomiques dont faisaient preuve les tumeurs puissent être la caractéristique essentielle du processus cancéreux. La découverte de grands remaniements chromosomiques conduisant à la création de gènes chimères dotés de propriétés nouvelles, véritables signatures de la pathologie cancéreuse, est venue conforter cette idée. On peut citer pour exemple la translocation (9;22), conduisant au fameux chromosome Philadelphie et dont le produit est la protéine de fusion BCR-ABL, pour la leucémie myéloïde chronique, ou les translocations impliquant le gène c-myc sur le chromosome 8 et les gènes d’immunoglobulines sur les chromosomes 2, 14 ou 22, pour les lymphomes de Burkitt (→). Dans cette perspective, la mutation est cependant un phénomène rare, mais qui constitue le moteur essentiel du processus. L’instabilité génétique, source de mutations, peut également être proposée comme moteur de la diversité présente au sein de la population des cellules cancéreuses. Cette position est confirmée par l’hypermutabilité observée dans de nombreuses tumeurs, hypermutabilité qui se décline au niveau nucléotidique, en ne mettant en jeu que des modifications locales (phénotype MIN), ou au niveau des chromosomes entiers (phénotype CIN). Les données acquises sur les mécanismes gouvernant l’agencement des deux phases essentielles que sont la duplication de l’ADN et la mitose éclairent notre compréhension du phénotype CIN. L’évolution de la tumeur peut alors se jouer dans le cadre des interactions entre cellules, qui vont faire émerger les clones des cellules les plus aptes à proliférer dans le micro-environnement tumoral.
(→) m/s 2003, n°2, p. 201
L’identification récente de BRCA-2 comme appartenant au groupe D2 de complémentation de l’anémie de Fanconi est de ce point de vue très illustratif. L’existence de points communs, au niveau moléculaire, entre la prédisposition au cancer du sein et l’anémie de Fanconi avait conduit Howlett et ses collaborateurs à séquencer les gènes BRCA- 1 et BRCA-2 dans les cellules appartenant aux groupes de complémentation B et D1 dont les gènes n’avaient pas encore été clonés [60, 61]. Des mutations sur les deux allèles de BRCA-2 ont été trouvées, qui dans tous les cas entraînent une perte de la région carboxy-terminale de la protéine. Cette situation est a priori surprenante au regard de la létalité observée chez les souris BRCA-2−/−. Cela peut s’expliquer par le fait que ne seront sélectionnés dans la tumeur que les allèles où seule la partie carboxy-terminale de la protéine est mutée, les autres mutants, conduisant à une diminution de la viabilité, étant rapidement éliminés. Cette observation explique parfaitement la différence de fréquence observée entre la prédisposition à l’anémie de Fanconi (< 1 pour 100000 naissances) et le pourcentage relativement élevé des hétérozygotes BRCA-2 (de l’ordre de 1 % dans la population américaine): il aura en effet fallu, dans le premier cas, un long processus de sélection des deux allèles ne conduisant pas à une létalité, alors que, dans le second cas, l’allèle sain « sauve la mise ». Ce constat est en tout cas édifiant pour les recherches futures, car de nombreux modèles murins, chez lesquels a été invalidé un gène de susceptibilité, développent des tumeurs dont ni la distribution tissulaire, ni le sous-type ne correspondent à ceux observés chez l’homme. Par exemple, alors que les mutations du gène p53 sont observées dans des tumeurs humaines d’origines très variées, les souris p53−/−développent majoritairement des lymphomes. De la même façon, les souris Rb+/- ne développent pas de rétinoblastome, mais meurent de tumeurs hypophysaires. La possibilité de réintroduire dans ces animaux (knock-in) des allèles présentant des mutations typiques des points chauds (zones où s’accumulent certaines mutations) observés dans les maladies humaines constitue de ce fait une perspective extrêmement intéressante.
Cette dernière remarque montre également la nécessité de se replacer dans un contexte intégré pour comprendre les aspects essentiels des mécanismes moléculaires responsables de l’évolution d’une tumeur. Ainsi, les travaux futurs devront prendre en compte un aspect important des processus cancéreux, celui de la perversion de l’espace et du temps cellulaire. Tout d’abord, les mécanismes concourant au bon déroulement de la phase S et de la mitose ne peuvent se comprendre que si l’on prend en compte les structures dans lesquelles ils se déploient. Par exemple, s’il est important de synthétiser, puis de détruire la cycline B au bon moment pour que la mitose se fasse correctement, encore faut-il que son ARNm soit traduit au bon endroit sur le fuseau mitotique [62]. De même, le temps cellulaire, tel qu’il est scandé par celui du cycle, n’est pas indépendant de celui de l’organisme. En effet, des données récentes du groupe de Cheng Chi Lee [63] montrent que des souris chez lesquelles le gène Period2, un régulateur clé du contrôle moléculaire des rythmes circadiens, a été muté, présentent une incidence accrue de cancers avec un dérèglement de l’expression de plusieurs régulateurs du cycle cellulaire et de la prolifération tels que les cyclines D1 et A et les protéines Mdm2 et c-myc.
Remerciements
Je remercie vivement Laure Coulombel, Bernard Ducommun et les collègues de l’IGMM et du CRBM pour leur lecture critique de ce manuscrit.
Références
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70 [Google Scholar]
- Fashema SJ, Thomas SM. Signalling by adhesion receptors. Nat Cell Biol 2000; 2: E225–36 [Google Scholar]
- Baron V, Lebrun P. Coopération entre les intégrines et les récepteurs à activité tyrosine kinase. Med Sci 2001; 17: 111–4 [Google Scholar]
- Rassoulzadegan M, Cowie A, Carr A, Glaichenhaus N, Kamen R, Cuzin F. The roles of individual polyoma virus early proteins in oncogenic transformation. Nature 1982; 300: 713–8 [Google Scholar]
- Le Peuch C, Dorée M. Le temps du cycle cellulaire. Med Sci 2000; 16: 461–8 [Google Scholar]
- Sherr CJ, Roberts JM. Cdk inhibitors: positive and negative regulators of G1- phase progression. Genes Dev 1999; 13: 1501–22. [Google Scholar]
- Sicinski P, Donaher JL, Parker SB, et al. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 1995; 82: 621–30. [Google Scholar]
- Tsutsui T, Heabi B, Moons DS, et al. Targeted disruption of Cdk4 delays cell cycle entry with enhanced p27Kip1 activity. Mol Cell Biol 1999; 19: 7011–9. [Google Scholar]
- Geng Y, Yu Q, Sicinska E, et al. Deletion of the p27Kip1 gene restores normal development in cyclin D1- deficient mice. Proc Natl Acad Sci USA 2001; 98: 194–9. [Google Scholar]
- Barnes EA, Kong M, Ollendorff V, Donoghue DJ. Patched 1 interacts with cyclin B1 to regulate cell cycle progression. EMBO J 2001; 20: 2214–23 [Google Scholar]
- Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol 2001; 2: 721–37. [Google Scholar]
- Motokura T, Bloom T, Goo- Kim H, et al. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature 1991; 350: 512–5. [Google Scholar]
- Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt EV. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature 1994; 369: 669–71. [Google Scholar]
- Bodrug SE, Warner BJ, Bath ML, Lindeman GJ, Arris AW, Adams JM. Cyclin D1 transgene impedes lymphocyte maturation and collaborates in lymphomagenesis with the myc gene. EMBO J 1994; 13: 2124–30. [Google Scholar]
- Lovec H, Grzeschiczk A, Kowalski MB, Moroy T. Cyclin D1/bcl-1 cooperates with myc genes in the generation of B-cell lymphoma in transgenic mice. EMBO J 1994; 13: 3487–95. [Google Scholar]
- Blanchard JM. Mécanismes moléculaires de la transformation oncogénique: quoi de neuf ? Bull Cancer 2002; 8 : 9–16. [Google Scholar]
- Fishel R, Wilson T. MutS homologs in mammalian cells. Curr Opin Genet Dev 1997; 7: 105–13. [Google Scholar]
- Kolodner R. Biochemistry and genetics of eukaryotic mismatch repair. Genes Dev 1996; 10: 1433–42. [Google Scholar]
- Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a MutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993; 75: 1215–25. [Google Scholar]
- Fishel R, Lescoe, MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993; 75: 1027–38. [Google Scholar]
- De Wind N, Dekker M, Berns A, Radman M, Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell 1995; 82: 321–30. [Google Scholar]
- Edelmann WE, Yang K, Umar A, et al. Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell 1997; 91: 467–77. [Google Scholar]
- De Wind N, Dekker M, Claij N, et al. HNPCC-like cancer predisposition in mice through simultaneous loss of Msh3 and Msh6 mismatch-repair protein functions. Nat Genet 1999; 23: 359–62. [Google Scholar]
- Wang Y, Cortez D, Yazdi P, et al. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 2000; 14: 927–39. [Google Scholar]
- Zhang H, Tombline G, Weber BL. BRCA1, BRCA2, and DNA damage response: collision or collusion? Cell 1998; 92: 433–6. [Google Scholar]
- Feunteun J. La prédisposition héréditaire au cancer du sein liée à BRCA1 et BRCA2: une maladie de la réponse aux lésions génotoxiques ? Med Sci 1999; 15: 38–44. [Google Scholar]
- Welcsh PL, Owens KN, King MC. Insights into the functions of BRCA1 and BRCA2. Trends Genet 2000; 16: 69–74. [Google Scholar]
- Wang Q, Zhang H, Fishel R, Greene MI. BRCA1 and cell signaling. Oncogene 2000; 19: 6152–8. [Google Scholar]
- Zheng L, Li S, Boyer TG, Lee WH. Lessons learned from BRCA1 and BRCA2. Oncogene 2000; 19: 6159–75. [Google Scholar]
- Bay JO, Uhrhammer N, Hall J, Stoppa-Lyonnet D, Bignon YJ. Fonctions de la protéine ATM et aspects phénotypiques de l’ataxietélangiectasie. Med Sci 1999; 15: 1086–95. [Google Scholar]
- Shiloh Y. ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev 2001; 11: 71–7. [Google Scholar]
- Gradia S, Acharya S, Fishel R. The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell 1997; 91: 995–1005. [Google Scholar]
- Fishel R. Mismatch repair, molecular switches, and signal transduction. Genes Dev 1998; 12: 2096–101. [Google Scholar]
- Fishel R. Signaling mismatch repair in cancer. Nat Med 1999; 5: 1239–41. [Google Scholar]
- Shih IM, Zhou W, Goodman SN, Lengauer C, Kinzler KW, Vogelstein B. Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res 2001; 61: 818–22. [Google Scholar]
- Jeanteur P. Le rôle d’APC dans la cancérogenèse colique: en plein dans le Myc! Bull Cancer 1998; 85: 925–8. [Google Scholar]
- Laurent-Puig P, Blons H. Mutations du gène APC et instabilité génétique. Med Sci 2001; 17: 954. [Google Scholar]
- Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer 2001; 1 : 55–67. [Google Scholar]
- Samowitz WS, Powers MD, Spirio LN, et al. β-catenin mutations are more frequent in small colorectal adenomas than in larger adenomas and invasive carcinomas. Cancer Res 1999; 59: 1442–4. [Google Scholar]
- Kaplan KB, Burds AA, Swedlow JR, Bekir SS, Sorger PK, Näthke IS. A role for the adenomatous polyposis coli in chromosome segregation. Nat Cell Biol 2001; 3: 429–32. [Google Scholar]
- Fodde R, Kuipers J, Rosenberg C, et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol 2001; 3: 433–8. [Google Scholar]
- Näthke IS, Adams CL, Polakis P, Sellin JH, Nelson WJ. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. J Cell Biol 1996; 134: 165–79. [Google Scholar]
- Mimori-Kiyosue Y, Shiina N, Tsukita S. Adenomatous polyposis coli (APC) protein moves along microtubules and concentrates at their growing ends in epithelial cells. J Cell Biol 2000; 148: 505–17. [Google Scholar]
- Peter M, Magnaghi-Jaulin L, Castro A, et al. Quand la dynamique chromosomique contrôle la division cellulaire. Pathol Biol 2001; 49: 649–54. [Google Scholar]
- Abrieu A, Dorée M. La cohésion des chromatidessoeurs et sa régulation au cours du cycle cellulaire. Med Sci 2001; 17: 353–4. [Google Scholar]
- Nigg E. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2001; 2:21–32. [Google Scholar]
- Gardner RD, Burke DJ. The spindle checkpoint: two transitions, two pathways. Trends Cell Biol 2000; 10: 154–8. [Google Scholar]
- Jallepalli PV, Lengauer C. Chromosome segregation and cancer: cutting through the mystery. Nat Rev Cancer 2001; 1: 109–17. [Google Scholar]
- Cahill DP, Lengauer C, Yu J, et al. Mutations of mitotic checkpoint genes in human cancers. Nature 1998; 392: 300–3. [Google Scholar]
- Gemma A, Seike M, Seike Y, et al. Somatic mutation of the hBUB1 mitotic checkpoint gene in primary lung cancer. Genes Chrom Cancer 2000; 29: 213–8. [Google Scholar]
- Michel LS, Liberal V, Chatterjee A, et al. MAD2 haplo-insufficiency causes premature anaphase and chromosomal instability in mammalian cells. Nature 2001; 409: 355–9. [Google Scholar]
- Kalitsis P, Earle E, Fowler KJ, Choo A. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev 2000; 14: 2277–82. [Google Scholar]
- Jallepalli PV, Waizenegger IC, Bunz F, et al. Securin is required for chromosomal stability in human cells. Cell 2001; 105: 445–7. [Google Scholar]
- Rousseau D. eIF-4E, régulation de la traduction et progression tumorale. Med Sci 2001; 17: 336–43. [Google Scholar]
- Hunter T. Signaling-2000 and beyond. Cell 2000; 100: 113–27. [Google Scholar]
- Takisawa H, Mimura S, Kubota Y. Eukaryotic DNA replication: from prereplication complex to initiation complex. Curr Opin Cell Biol 2000; 12: 690–6. [Google Scholar]
- Kelly TJ, Brown GW. Regulation of chromosome replication. Annu Rev Biochem 2000; 69: 829–80. [Google Scholar]
- Bulavin DV, Amundson SA, Fornace Jr AJ. P38 and Chk1 kinases: different conductors for the G2/M checkpoint symphony. Curr Opin Genet Dev 2002; 12: 92–7. [Google Scholar]
- Scheid MP, Woodgett JR. PKB/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol 2001; 2: 760–8. [Google Scholar]
- Howlett NG, Tanigushi T, Olson S, et al. Biallelic inactivation of BRCA-2 in Fanconi anemia. Science 2002; 297: 606–9. [Google Scholar]
- Jeanteur P. L’anémie de Fanconi et les gènes BRCA: même combat ? Bull Cancer 2002; 89: 917–8 [Google Scholar]
- Groisman I, Huang YS, Mendez P, Cao Q, Theurkauf W, Richter JD. CPEB, Maskin, and cyclin B1 mRNA at the mitotic apparatus: implications for local translational control of cell division. Cell 2000; 103: 435–47. [Google Scholar]
- Fu L, Pelicano H, Liu J, Huang P, Lee CC. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 2002; 111: 41–50. [Google Scholar]
Liste des figures
|
Figure 1. Le paradigme de la signalisation via les MAP-kinases et de sa coopération avec les intégrines. La fixation d’un ligand sur un récepteur doué d’activité tyrosine kinase conduit à la multimérisation de ce dernier et à son activation par autophosphorylation. Les tyrosines ainsi phophorylées au sein d’une courte séquence d’acides aminés constituent autant de cibles pour des protéines possédant des domaines susceptibles de les reconnaître, les domaines SH2 (Src homology domain 2) étant les plus représentatifs [55]. La fixation du ligand crée ainsi une surface d’interaction conditionnelle à la phosphorylation qui va alors recruter directement un effecteur ou passer par l’intermédiaire d’une molécule adaptatrice. L’activation de Ras est une parfaite illustration de ce dernier cas: il est ancré dans la membrane plasmique via un groupement farnésyl carboxy-terminal, et se trouve recruté par le récepteur via le complexe formé entre Grb2 et Sos. Grb2 est constitué d’un domaine SH3, associé à Sos de façon constitutive, et de deux domaines SH2, qui fixent des tyrosines phosphorylées dans des contextes appropriés. Ce faisant, Ras est mis en présence du facteur d’échange qu’est Sos, et passe de la forme se liant au GDP à celle se liant au GTP, active et capable d’interagir avec de nombreux effecteurs, dont Raf-1. Celui-ci constitue le premier terme d’une cascade de kinases constituée de trois modules, MAPKKK (Raf-1), MAPKK (MEK) et MAPK (ERK), cette dernière pouvant passer dans le noyau et phosphoryler des facteurs de transcription spécifiques. L’interaction de certaines intégrines avec la matrice extracellulaire peut également recruter les mêmes complexes et participer ainsi à l’activation de Ras. Les intégrines ne possédant pas d’activité kinase, plusieurs mécanismes peuvent être envisagés. Un mécanisme complexe, impliquant les kinases FAK (focal adhesion kinase) et src dans un jeu réciproque de phosphorylations, qui vont attirer une protéine adaptatrice telle que Grb2, est vraisemblablement impliqué dans la motilité cellulaire. Toutefois, la signalisation mitogène passerait par le recrutement de l’adaptateur Shc via la protéine membranaire cavéoline (Cav), la phosphorylation s’effectuant par l’un des membres de la famille Src. Quelques exemples parmi les premiers oncogènes viraux isolés: des facteurs de croissance comme sis (chaîne B du PDGF) ou int-2 (un variant du FGF); des kinases membranaires comme src, yes, lck, ou erbB (version tronquée du récepteur de l’EGF); des protéines G comme H-ras ou K-ras; des kinases cytoplasmiques comme raf/mil, mos ou pim-1; des facteurs de transcription comme myc, jun, fos ou erbA (récepteur de l’hormone thyroïdienne T3). |
| Dans le texte | |
|
Figure 2. Contrôle des transitions G1/S et G2/M. A. Les origines de réplication (rectangles jaunes) fixent pendant la phase G1 le complexe hexamérique ORC (origin recognition complex), qui sert à son tour de plate-forme sur laquelle viennent se loger, de façon séquentielle, Cdc6, Cdt1, le complexe hexamérique MCM (mini-chromosome maintenance) (hélicase ?), et les facteurs requis pour le démarrage proprement dit de la réplication: Cdc45, RFA (replication factor A) et l’ADN polymérase α [56, 57]. L’assemblage et l’activation d’un tel complexe sont sous la dépendance, d’une façon qui n’est pas encore claire, des kinases Cdk2-cycline E et Cdc7-Dbf4. Les protéines Cdc6 et Cdt1 joueraient un rôle essentiel en recrutant ces facteurs afin de constituer le complexe de pré-initiation. Quand la réplication est enclenchée, Cdc6 et Cdt1 se détachent de la chromatine, à la suite d’une phosphorylation de Cdc6, vraisemblablement par Cdk2-cycline A. Cdc6 phosphorylé est exporté vers le cytoplasme. Quant à Cdt1, il est probablement inactivé par interaction avec la géminine. La reconstitution d’un complexe de préinitiation sur les origines qui ont été activées est ainsi évitée, et il ne peut, de ce fait, y avoir à nouveau réplication. Par ailleurs, sont également associées (mais non montrées sur ce schéma) des protéines de surveillance comme ATR (voir plus loin) qui vont arrêter la réplication en cas d’apparition de défauts au niveau de la fourche de réplication. B. Le complexe Cdk1/cycline B se forme à la suite de la synthèse de la cycline B en fin de phase S et de sa translocation depuis le cytoplasme (phases S et G2) vers le noyau (début de mitose). Son activité est contrôlée par une série de phosphorylations antagonistes. À la phosphorylation activatrice par la CAK (Cdk activating kinase) sur la thréonine 161 (boucle T, module l’interaction avec la cycline), s’opposent les phosphorylations inhibitrices sur la thréonine 14 et la tyrosine 15 (site de liaison de l’ATP) par les kinases Wee1 et Myt1 ([46] et Y. Pommier et K.W. Kohn, p.173 de ce numéro). La mutation de ces deux derniers acides aminés produit une entrée prématurée en mitose. Les résidus sérine 14 et tyrosine 15 sont déphosphorylés par les phosphatases Cdc25B et Cdc25C. L’activité et la localisation de ces phosphatases seraient également soumises à régulation, après phosphorylation par les kinases p38 pour Cdc25B, et Chk1 et Chk2 pour Cdc25C. Cette dernière, nucléaire juste avant la mitose, serait maintenue dans le cytoplasme à l’interphase, en raison de son interaction avec une protéine 14-3-3, après sa phosphorylation sur la sérine 216 par Chk1 et Chk2. Cdc25B contient plusieurs sites de fixation des protéines 14-3-3, dont la sérine 309, qui serait ciblée par la kinase de stress p38 en présence de lésions induites par les rayonnements ultraviolets [58]. Plk1 (Polo-like kinase) interviendrait également en amont et participerait à l’activation de Cdc25c. Quant aux autres phosphatases, elles doivent pour l’essentiel être caractérisées, PP2A supprimant vraisemblablement les phosphorylations apportées par les Cdk. La présence de lésions dans l’ADN active également une autre voie qui aboutit à l’inhibition de l’activité du complexe Cdk1-cycline B: l’induction de l’inhibiteur p21 par p53. |
| Dans le texte | |
|
Figure 3. La production de la cycline D1, depuis sa synthèse jusqu’à sa dégradation, est sous le contrôle de stimulus externes. L’expression de la cycline D1 aurait pour finalité de faire sortir les cellules de leur état de quiescence et de rendre le cycle cellulaire dépendant de l’exposition des cellules à des facteurs de croissance, l’induction de la cycline E étant l’enjeu de ce processus. Ce dernier point est illustré par les expériences d’invalidation des gènes codant pour la cycline D1 ou la kinase Cdk4. Les souris déficientes en cycline D1 ou en Cdk4 viennent à terme, mais avec des problèmes de développement tels qu’une déficience oculaire, une taille réduite et une absence de maturation lobulo-alvéolaire terminale des glandes mammaires [7, 8]. Les animaux cycline D1−/−, chez lesquels la cycline E est réexprimée après insertion de son gène dans le locus cycline D1 (knock-in), ont un phénotype quasi normal. L’expression du gène cycline E est alors directement sous la dépendance des signaux extrinsèques, et ne nécessite plus une activation préalable de la cycline D1. Dans le même ordre d’idées, l’invalidation, dans les fonds génétiques cycline D1−/− ou Cdk4−/−, du gène codant pour p27Kip1, inhibiteur du complexe Cdk2-cycline E, supprime partiellement ces désordres [9]. Ce dernier résultat illustre le double rôle que joue le complexe Cdk4-cycline D1: activer la transcription du gène cycline E, et limiter la quantité libre de l’inhibiteur p27Kip1, avec pour résultat net une augmentation de l’activité Cdk2-cycline E. L’accumulation de la cycline D1 résulte, d’une part, de l’induction transcriptionnelle de son gène, et, d’autre part, de l’activation de la traduction de son ARN messager. Ces deux événements sont sous le contrôle de Ras via deux voies différentes: Raf/Mek/Erk et PI3-K/Akt/GSK-3β. Cette dernière voie contrôlerait également la localisation subcellulaire et la dégradation de la cycline D1. GSK-3β phosphoryle la thréonine 286 de la cycline qui est alors redirigée, pendant la phase S, vers le cytoplasme où elle est ubiquitinylée et dégradée par le protéasome. Akt, homologue cellulaire de l’oncogène v-Akt présent dans le virus AKT8, appartient à une famille de protéine kinases activées par les phospholipides engendrés par la PI-3K [59]. Elle voit donc son activité contrôlée par le suppresseur de tumeur PTEN (phosphatase and tensin homolog deleted on chromosome ten), capable d’hydrolyser les phospho-inositides. Elle est également phosphorylée par l’ILK (integrin linked kinase), liant ainsi la dégradation de la cycline D1 à l’adhérence cellulaire. |
| Dans le texte | |
|
Figure 4. Interaction entre le suppresseur de tumeur Ptc et la cycline B1. La nævomatose basocellulaire, ou syndrome de Gorlin, est une maladie autosomique dominante prédisposant à l’apparition de nombreux cancers et anomalies du développement. Le gène de susceptibilité à ce syndrome a été localisé en 9q22.3, et la présence de déséquilibres alléliques sur ce site, observés dans les carcinomes basocellulaires aussi bien familiaux que sporadiques, en fait un gène suppresseur de tumeurs dont le mode de fonctionnement est encore inconnu. Des résultats récents montrent que Ptc peut interagir avec la cycline B1 [10]. Celle-ci est localisée dans le cytoplasme pendant les phases S et G2, puis dans le noyau en début de mitose, vraisemblablement grâce à une interaction avec la cycline F. Une séquence d’acides aminés en position amino-terminale, initialement caractérisée comme séquence de rétention cytoplasmique, fait office de séquence d’export nucléaire (SEN) qui, sous sa forme non phosphorylée (sérines 94, 96, 101, 113), permet son retour dans le cytoplasme par l’intermédiaire de CRM1 (chromosomal maintenance region 1). Plusieurs kinases sont susceptibles de phosphoryler le SEN: Cdk1-cycline B (Ser 94 et Ser 96) et Plk1 (Ser 113). La forme phosphorylée de la cycline B1 interagit avec Ptc en l’absence de son ligand, Shh, l’ajout de ce dernier provoquant la rupture de cette association. Ptc interagit aussi avec Smo (Smoothed) - une autre protéine trans-membranaire - qui serait en fait le composant chargé de la signalisation en aval. Cette observation ouvre ainsi deux possibilités non exclusives. D’une part, la rétention de la cycline B1 par Ptc dans le cytoplasme permet de retarder la mitose en l’absence de Shh, ce qui expliquerait le caractère suppresseur de tumeurs de Ptc. D’autre part, comme Smo est phosphorylé après liaison de Shh sur Ptc, qui n’est pas une kinase, le complexe Cdk1-cycline B1 pourrait être responsable de cette phosphorylation (cela n’est toutefois encore que pure spéculation). |
| Dans le texte | |
|
Figure 5. BASC, un complexe de surveillance de l’intégrité de l’ADN ? BASC (BRCA1- associated genome surveillance complex) [24] est constitué par une quarantaine de protéines déjà connues pour s’associer à BRCA1, telles BARD1 (BRCA1-associated ring domain) ou HDAC1 (histone deacetylase 1), ainsi que par des protéines impliquées dans la détection de lésions, comme le complexe MSH2-MSH6 ou encore ATM, et dans la recombinaison homologue, comme le complexe RAD50-MRE11-NBS1. Sont également présentes les trois sous-unités du facteur de réplication RFC impliqué dans la mise en place de PCNA (proliferating cell nuclear antigen, un co-facteur de l’ADN polymérase δ) sur la fourche de réplication. Pour le moment, on ne sait pas si BASC est constitué d’un seul complexe, ou de plusieurs sous-complexes dévolus à des tâches différentes. De même, le rôle de MSH2-MSH6 est ouvert: soit il est impliqué dans le recrutement de BASC après avoir reconnu la lésion, soit il est toujours associé à BASC, et c’est un changement de conformation, lié à l’hydrolyse de l’ATP, qui joue un rôle dans la signalisation (ces deux hypothèses n’étant pas exclusives) [24–30]. Les gènes MSH ont été invalidés chez la souris et les cellules issues de ces animaux montrent une instabilité des satellites, une tolérance vis-à-vis des agents alkylants et, de façon intéressante, un laxisme de la recombinaison homologue visà- vis de l’identité des séquences d’ADN impliquées. Les animaux MSH2−/− ne présentent pas d’anomalies majeures, mais 30 % d’entre eux développent des lymphomes à un âge précoce [21]. De même, les souris MSH6−/− développent toutes des lymphomes et des tumeurs cutanées ou utérines, cette distribution pouvant changer avec le fond génétique des animaux utilisés, suggérant ainsi que les complexes MSH2/MSH6 et MSH2/MSH3 ont des spécificités différentes in vivo. |
| Dans le texte | |
|
Figure 6. Voie de signalisation Wnt/Frizzled/caténine β. A.La caténine β est soit libre, soit engagée dans un complexe avec la caténine β et les cadhérines au niveau des jonctions adhérentes. En l’absence de Wnt, la caténine β libre est dégradée par le protéasome, via un complexe de destruction contenant APC, l’axine et la glycogène-synthétase-kinase-3 β(GSK-3 β). Cette dernière phosphoryle la caténine β, reconnue alors par le complexe SCF (Skip1- Culin-Fbox) via la protéine F-box β-TrcP, ce qui entraîne son ubiquitinylation et sa dégradation. Ce faisant, la transcription des gènes contrôlés par LEF/Tcf (lymphoid-enhancer factor/T-cell factor) est silencieuse, ce dernier étant inhibé dans le noyau par le co-répresseur Groucho. B. En présence de WNT, le récepteur Frizzled, associé à une protéine apparentée au récepteur des lipoprotéines de basse densité (LRP6), est activé. S’ensuit une cascade d’événements peu documentée au cours de laquelle l’inhibiteur de GSK-3β GBP est activé via Dishevelled (pour plus de détails, voir [38]). En conséquence, la caténine β n’est plus dégradée, diffuse dans le noyau, déplace Groucho et active la transcription sous le contrôle de LEF/Tcf. APC contrôle ainsi le destin intracellulaire de la caténine β en orchestrant sa distribution entre les jonctions adhérentes, le cytoplasme et le noyau. |
| Dans le texte | |
|
Figure 7. APC et APC/C se retrouvent dans les kinétochores. APC (adenomatous polyposis coli) et APC/C (anaphase promoting complex) se retrouvent au niveau des kinétochores, structures chargées du lien entre les chromosomes et les microtubules constituant le fuseau mitotique (flèches vertes épaisses). APC est soit impliqué dans le contrôle, soit est un acteur essentiel de la dynamique des microtubules. Il est phosphorylé, au moins in vitro, par les kinases Bub1-Bub3 et BubR1-Bub3. Celles-ci sont vraisemblablement également impliquées dans le recrutement de Mad2 par Mad1 ou APC/C, ce dernier étant inactif tant que tous les kinétochores ne sont pas engagés dans une liaison et que tous les chromosomes ne sont pas correctement alignés (A). Quand cela se produit, un signal encore mal identifié (structure de la jonction microtubule-kinétochore ? tension du fuseau sur le kinétochore ?) est interprété via CENP-E (centromeric protein E) en termes de phosphorylations par les kinases Bub1-Bub3, BubR1-Bub3 et Mps1. Mad2 se dissocie alors de APC/C, qui est ainsi activé (B). Cdc20/Fizzy/p55CDC est une protéine adaptatrice qui permet à APC/C de diriger la sécurine vers le protéasome après son ubiquitinylation, ce qui a pour effet d’activer la séparase, protéase responsable de la dégradation des cohésines. Ces dernières forment un complexe d’au moins quatre sous-unités, responsable de l’attachement entre les chromatides soeurs. Plusieurs autres kinases interviennent très vraisemblablement. Ainsi, lors d’une lésion dans l’ADN, Chk1 phosphoryle la sécurine, ce qui la rend insensible à APC/C. Mais l’on trouve également au niveau du kinétochore MAPK activée, Plk1 et Aurora B, qui semblent également intervenir dans la cytodiérèse, mais pour lesquelles il est encore difficile de définir un rôle exact. Les gènes Plk1, Aurora A, Aurora B et Aurora C sont associés à des locus chromosomiques altérés dans de nombreux cancers. Aurora A et Plk1 sont surexprimées dans certaines tumeurs primaires, et sont capables de transformer des cellules en culture. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.