")
")
| Issue |
Med Sci (Paris)
Volume 36, Décembre 2020
Les Cahiers de Myologie
|
|
|---|---|---|
| Page(s) | 22 - 27 | |
| Section | Dystrophies musculaires des ceintures (LGMD) | |
| DOI | https://doi.org/10.1051/medsci/2020243 | |
| Published online | 11 January 2021 | |
Les sarcoglycanopathies
État des lieux et perspectives thérapeutiques
Sarcoglycanopathies: state of the art and therapeutic perspectives
1
Centre de Référence des maladies neuromusculaires Nord/Est/Île-de-France, APHP, Groupe Hospitalier Pitié-Salpêtrière, Sorbonne Université, Paris, France.
2
Laboratoire de biochimie génétique. APHP, Hôpital Cochin, Paris, France.
3
Centre de Référence des maladies neuromusculaires Nord/Est/Île-de-France. APHP, CHU Raymond Poincaré, Garches. Université Paris-Saclay, France.
4
Généthon, 91000, Évry, France
5
Université Paris-Saclay, Université d’Evry, Inserm, Généthon, unité de recherche Integrare UMR_S951, 91000, Évry, France
* This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les sarcoglycanopathies font partie des dystrophies musculaires des ceintures (LGMD) autosomiques récessives et représentent la troisième cause la plus fréquente d’entre elles. Elles sont consécutives à un déficit d’un des sarcoglycanes α, β, γ, ou δ. La présentation clinique habituelle est celle d’une atteinte symétrique des muscles des ceintures pelvienne et scapulaire ainsi que du tronc, associée à une atteinte cardiorespiratoire plus ou moins sévère et une élévation franche des créatine-phospho-kinases (CPK). Les premiers symptômes apparaissent au cours de la première décennie, la perte de la marche survenant souvent au cours de la deuxième décennie. Les lésions sont de type dystrophique sur la biopsie musculaire. Il s’y associe une diminution ou une absence d’immunomarquage du sarcoglycane correspondant au gène muté, et dans une moindre mesure des trois autres sarcoglycanes associés. De nombreuses mutations ont été rapportées dans les quatre gènes impliqués et quelques-unes d’entre elles sont prépondérantes dans certaines populations. à ce jour, il n’existe pas de traitement curatif ce qui n’empêche pas de voir se développer de nombreux essais cliniques, notamment en thérapie génique.
Abstract
Sarcoglycanopathies are the third most common cause of autosomal recessive limb girdle muscular dystrophies (LGMD). They are the result of a deficiency in one of the sarcoglycans a, b, g, or d. The usual clinical presentation is that of a symmetrical involvement of the muscles of the pelvic and scapular girdles as well as of the trunk, associated with more or less severe cardio-respiratory impairment and a marked increase of serum CK levels. The first symptoms appear during the first decade, the loss of ambulation occurring often during the second decade. Lesions observed on the muscle biopsy are dystrophic. This is associated with a decrease or an absence of immunostaining of the sarcoglycan corresponding to the mutated gene and, to a lesser degree, of the other three sarcoglycans. Many mutations have been reported in the four incriminated genes and some of them are prevalent in certain populations. To date, there is no curative treatment, which does not prevent the development of many clinical trials, especially in gene therapy.
© 2020 médecine/sciences – Inserm
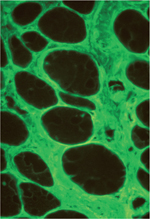
© Inserm/Hantaï, Daniel
Les sarcoglycanopathies font partie du groupe des dystrophies musculaires des ceintures (LGMD pour limb girdle muscular dystrophy) autosomiques récessives et sont causées par des mutations dans des glycoprotéines transmembranaires appelées sarcoglycanes (SG) [1]. Les sarcoglycanes (SG) alpha (α), bêta (β), gamma (γ) et delta (δ), font partie du complexe glycoprotéique associé à la dystrophine (DGC). La dystrophine et le DGC, via une liaison du cytosquelette d’actine à la matrice extracellulaire, confère une réelle stabilité au sarcolemme et protège les fibres musculaires striées et cardiaques de l’apparition de lésions induites par la contraction musculaire [2, 3]. On dénombre quatre gènes associés aux dystrophies des ceintures : SGCA (correspondant à la LGMD-R3), SGCB (pour la LGMD-R4), SGCG (pour la LGMD-R5) et SGCD (pour la LGMD-R6), et qui codent respectivement les protéines sarcoglycanes de type alpha, bêta, delta et gamma (Figure 1). Deux autres sarcoglycanes, nommés epsilon (codé par le gène SGCE) et zêta (codé par le gène SGCZ), ne sont pas associées à des maladies musculaires. Le gène SGCE peut entraîner un tableau de dystonie héréditaire avec myoclonies. Quant au gène SGCZ, il n’a été incriminé, à ce jour, dans aucune maladie génétique humaine [4].
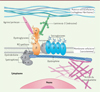 |
Figure 1. Les quatre sarcoglycanes (α, β, γ, δ) font partie du complexe DGC associé à la dystrophine. © AFM-Téléthon. |
Épidémiologie (Tableau I)
Les sarcoglycanopathies représentent la troisième cause la plus fréquente de LGMD de transmission autosomique récessive après les calpaïnopathies et les dysferlinopathies [5, 6]. La prévalence estimée est de 3,4/100 000 pour le SGCA, 0,8/100 000 pour le SGCB, 0,1/100 000 pour le SGCG et 0,07/100 000 pour le SGCD ; mais elle est très variable à travers le monde. En France, on estime la prévalence à environ 400 patients atteints de sarcoglycanopathies génétiquement confirmées. Les mutations du SGCA sont les plus répandues en Europe [7], tandis que les mutations du SGCG sont les plus courantes dans la population nord-africaine et tzigane [8]. Les mutations du SGCB sont ubiquitaires [7, 9-11], les mutations dans le SGCD restent exceptionnelles et semblent cantonnées à la population brésilienne [12]. De nombreuses mutations ont été rapportées mais on notera que certaines mutations sont particulièrement prépondérantes, comme la mutation homozygote c.525delT (p.Phe175Leufs*20) du gène SGCG (observées chez les patients d’Afrique du Nord et du pourtour méditerranéen) ou la mutation c.229C>T (p.Arg77Cys) retrouvée à l’état homozygote ou hétérozygote composite dans le gène SGCA [1, 7, 13].
Caractéristiques épidémiologiques et cliniques des sarcoglycanopathies (d’après le site web neuromuscular.wustl.edu et [11, 24]).
Clinique
Les sarcoglycanopathies puis les gènes impliqués ont été identifiés dans les années 1990 [14] et depuis, plusieurs études ont essayé de préciser les caractéristiques phénotypiques et génotypiques de chacune d’entre elles [11, 15-17]. La présentation clinique des sarcoglycanopathies est celle d’une LGMD caractérisée par une atteinte symétrique des muscles des ceintures pelvienne et scapulaire ainsi que du tronc, avec divers degrés d’atteinte cardiorespiratoire [11, 16, 18, 19]. Un décollement des omoplates, une pseudohypertrophie des mollets et de la langue, ainsi que des CPK très élevées (> 1 000 U/l) complètent typiquement le tableau clinique (Figure 2).
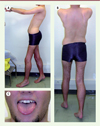 |
Figure 2. Cas de sarcoglycanopathie-alpha chez un jeune homme de 25 ans présentant une dystrophie musculaire des ceintures évoluant lentement depuis l’enfance. On notera la pseudo-hypertrophie des mollets (A, B), l’hyperlordose (A), les rétractions achilléennes (A, B), le décollement des omoplates (B) et la discrète macroglossie (C). Deux mutations hétérozygotes ont été identifiées dans le gène SGCA (une mutation d’épissage dans l’intron 6 et une mutation dans l’exon 3, la p.R77C). |
Dans les formes les plus sévères, elles peuvent mimer en tout point une dystrophie musculaire de type Duchenne. Dans les formes moins sévères, elles peuvent être confondues avec une dystrophie musculaire de Becker. Des phénotypes plus rares, tels qu’une hyperCKémie peu ou symptomatique, ou une intolérance à l’effort, ont également été rapportés [20]. Une scoliose est présente chez 33 à 48 % des patients, indépendamment du gène concerné, et des rétractions musculo-tendineuses apparaissaient fréquemment au cours de l’évolution de la maladie. On notera qu’il est quasi-impossible de faire la distinction, au niveau clinique, entre les quatre sous-types de sarcoglycanopathie.
Évolution
Le début des symptômes, telle que la faiblesse des membres inférieurs, se situe dans la première décennie. Il est plus précoce pour les patients γ-SG que α-SG [11]. Dans une grande majorité de cas, la perte de la marche est observée au cours de l’adolescence ou au cours de la deuxième décennie [11]. L’atteinte des muscles cardiaque et respiratoires fait souvent partie du tableau clinique [21-23]. La présence d’une cardiomyopathie dilatée est plus élevée dans les β-SG (20-26 %) et les γ-SG (12-37 %) tandis que celle-ci est moins fréquente dans les a-SG (0,6-11 %). Une insuffisance respiratoire restrictive est fréquente menant dans 23-29 % des cas à une ventilation non invasive et plus rarement, dans 2 % des cas, à une trachéotomie [11]. La diminution de la capacité vitale est corrélée avec la perte de la marche [24].
Diagnostic
Le diagnostic de certitude de sarcoglycanopathie repose sur l’analyse des quatre gènes des sarcoglycanes, soit par séquençage Sanger (analyse gène par gène), soit par NGS (Next Generation Sequencing), en utilisant un panel plus ou moins large de gènes neuromusculaires (panel LGMD par exemple), ou en explorant l’exome entier. En amont, la biopsie musculaire, si elle est réalisée, permet déjà d’orienter le diagnostic moléculaire en montrant l’absence ou une réduction d’immununomarquage d’un des sarcoglycanes [7-25] et de façon secondaire, une diminution de signal d’un ou plusieurs des sarcoglycanes non mutés, voire même de la dystrophine elle-même. Ces anomalies de signal peuvent être difficiles à interpréter et sont parfois sources de confusion. Devant un phénotype clinique évocateur de sarcoglycanopathie chez un patient originaire d’Afrique du Nord ou appartenant à la communauté tzigane, la réalisation d’une biopsie musculaire n’est pas toujours nécessaire, et une analyse en première intention des deux mutations causales (la mutation c.525delT du gène SGCG [24], la c.848G>A (p.Cys283Tyr)[26]). Des défauts quantitatifs comme des délétions d’exons entiers peuvent aussi être mis en évidence.
Prédicteurs de progression
Une étude européenne récente a montré que les mutations conduisant à l’absence ou à un taux de moins de 30 % de l’expression d’une des protéines sont associées à un phénotype plus sévère, avec une perte de la marche survenant avant l’âge de 18 ans [11]. La précocité de l’âge du début des symptômes est corrélée avec une évolutivité plus marquée de la maladie [11]. Globalement, les patients porteurs de mutations sur les gènes SGCB et SGCG ont un phénotype plus sévère et une perte de la marche plus précoce que les patients ayant des mutations du gène SGCA [11]. L’analyse des mutations peut être utile pour prédire l’expression de la protéine dans de nombreuses maladies. En général, la présence de deux mutations tronquées est associée à une perte totale ou sévère de l’expression des protéines [27, 28]. Néanmoins, les corrélations génotypes-phénotypes restent difficiles à établir, une mutation faux-sens pouvant créer un site d’épissage cryptique ou une dégradation des sarcoglycanes lors de leur passage dans le réticulum endoplasmique. Par conséquent, certaines mutations faux sens peuvent être à l’origine d’une très faible expression de la protéine voire d’une absence complète de l’expression de celle-ci [29, 30].
Perspectives thérapeutiques
Les mutations des sarcoglycanes sont de nature autosomique récessive et correspondent à une perte de fonction de la protéine. Une approche thérapeutique par remplacement du gène est donc adaptée. A ce jour, cette stratégie pour les maladies neuromusculaires est réalisée par un transfert de gène médié par un vecteur dérivé du virus adéno-associé (AAV). De nombreuses preuves de principe de l’efficacité d’une telle stratégie visant à restaurer le phénotype de souris déficientes en sarcoglycanes ont été rapportées [31-46]. Ces données précliniques ont été à la base de plusieurs essais cliniques avec différents modes d’administration (injection intramusculaire, perfusion de membres ou injection systémique). On remarquera que les essais utilisant une approche locale dans les sarcoglycanopathies ont été parmi les premiers essais de thérapie génique mis en œuvre dans les maladies neuromusculaires.
Concernant les approches thérapeutiques par administration intramusculaire : deux essais de phase I utilisant le sérotype AAV1 ont été menés, l’un ciblant le sarcoglycane a (NCT00494195) et l’autre le sarcoglycane g (NCT01344798). L’essai ciblant le sarcoglycane a était promu par le Nationwide Children’s Hospital (Ohio, USA), le Dr Jerry Mendell en étant l’investigateur principal, et s’est déroulé entre 2008 et 2011. Il a consisté à injecter dans le muscle extensor digitorum brevis un vecteur exprimant l’ADN complémentaire codant le sarcoglycane a humain sous la dépendance du promoteur dérivé de la créatine kinase (tmck) à la dose de 3,25 × 10¹¹ vg. Deux cohortes ont été analysées : l’une avec une biopsie réalisée précocement sur deux patients à 45 jours et sur un patient à 90 jours, et l’autre plus tardive avec une biopsie réalisée après 6 mois chez trois patients. Les patients ont tous reçus un traitement préalable comportant de la methylprednisolone. Les résultats de la première cohorte ont montré une augmentation de 4 à 5 fois de l’expression de la sarcoglycane a, une restauration du complexe sarcoglycane et une augmentation de la taille des fibres musculaires [47]. Aucun effet indésirable majeur n’a été noté. Des anticorps neutralisants contre l’AAV ont été observés dès la deuxième semaine après l’injection. Les résultats de la deuxième cohorte [39] ont montré une expression persistante du sarcoglycane a chez deux sujets sur trois. Il a été démontré que le patient avec le résultat négatif avait développé des anticorps neutralisants et une réponse cellulaire T dès le deuxième jour après injection. L’essai concernant le sarcoglycane g, quant à lui, était promu par Généthon et s’est déroulé entre 2006 et 2011, avec le Pr Serge Herson (Groupe Hospitalier Pitié-Salpêtrière) comme investigateur principal. L’essai a consisté à injecter dans le muscle extenseur radial du carpe, et à doses croissantes (3,0 × 1010 vg, 1,5 × 1010 vg, et à 4,5 × 1010 vg en deux injections, sur 3 cohortes de 3 patients), un vecteur exprimant l’ADN complémentaire codant le sarcoglycane g humain, sous la dépendance d’un promoteur dérivé de la desmine (AAV1-des-hγSGC). Les résultats obtenus trente jours après l’injection ont permis de détecter le vecteur chez tous les patients sauf un ainsi que la présence de la protéine par immunohistochimie, et la reconstitution du complexe sarcoglycane dans le troisième groupe [48]. Aucun effet indésirable majeur n’a été observé. Les patients sont tous devenus séropositifs pour le vecteur AAV1 et un patient a déclenché une réponse cytotoxique contre la capside du vecteur.
Un essai avec une approche par perfusion de membres a été réalisé pour le sarcoglycane (NCT01976091) entre 2015 et 2019. Le produit injecté (SRP-9004) était un vecteur de sérotype rh74 exprimant l’ADN complémentaire codant le sarcoglycane a, sous la dépendance du promoteur tmck (scAAVrh74.tMCK.hSGCA). La particularité de ce vecteur est qu’il est double brin, par complémentarité des brins. Ce produit a été licencié à Myonexus Therapeutics, une biotech rachetée par le laboratoire pharmaceutique Sarepta Therapeutics en 2019. Cet essai de phase I/II avec escalade de dose était également sous la responsabilité du Dr Jerry Mendell. Six patients ont été injectés sur un total de 9 patients prévu initialement. Un premier patient adulte non-ambulant a été injecté dans l’artère fémorale à la dose de 1E12 vg/kg dans un membre puis cinq patients âgés de 8 à 13 ans ont reçu de façon bilatérale soit 1E12 vg/kg/membre soit 3E12 vg/kg/membre. Les résultats ont montré à six mois post-injection la présence du vecteur et de la protéine avec une augmentation de force objectivée chez deux patients mais ne s’accompagnant pas d’une amélioration notable du test de marche (6MWT) [49].
Tout aussi intéressant, un premier essai de phase I/II (NCT03652259) utilisant une administration systémique d’un produit de thérapie génique a débuté en octobre 2018 pour le sarcoglycane b. Cet essai sponsorisé par le laboratoire Sarepta Therapeutics a pour objectif d’évaluer à différentes doses un AAV de sérotype rh74 exprimant l’ADN complémentaire codant le sarcoglycane b humain, sous la dépendance du promoteur MHCK7 (scAAVrh74.MHCK7.hSGCB ; SRP-9003). Cet essai doit se terminer en décembre 2020 mais des résultats intermédiaires très encourageants ont déjà été annoncés par Sarepta Therapeutics. Six patients âgés entre 4 et 13 ans ont été injectés avec deux doses : soit 5E13 vg/kg, soit 1E14 vg/kg. Les biopsies musculaires prélevées à 90 jours montrent une bonne expression de la protéine (environ 36 % pour la première cohorte et 62 % pour la deuxième). Les évaluations fonctionnelles montrent une amélioration avec une augmentation de 5,7 % sur l’échelle NSAD (North Star Assessment for Dysferlinopathy) 18 mois après injection pour le première cohorte et 3,7 % à 6 mois pour la deuxième cohorte. Les effets indésirables ont été modérés voire minimes.
Essais cliniques dans les sarcoglycanopathies.
En conclusion, les résultats obtenus dans ces essais cliniques sont très encourageants et montrent l’intérêt de poursuivre dans cette voie de thérapie génique. De prochains essais devraient voir le jour prochainement puisque Sarepta Therapeutics a un programme incluant les sarcoglycanes b, a et g. Généthon, pour sa part, prépare un essai en administration systémique pour le sarcoglycane g dans un futur très proche, puis un deuxième ciblant le sarcoglycane a.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
À Célia Thevenard (Généthon) pour le travail de recherche bibliographique et la génération du tableau de la section thérapeutique.
Références
- Duggan DJ, Gorospe JR, Fanin M, et al. Mutations in the sarcoglycan genes in patients with myopathy. N Engl J Med 1997 ; 336 : 618–624. [Google Scholar]
- Gao QQ, McNally EM. The dystrophin complex: structure, function, and implications for therapy. Compr Physiol 2015 ; 5 : 1223–1239. [Google Scholar]
- Nigro V, Savarese M. Genetic basis of limb-girdle muscular dystrophies: The 2014 update. Acta Myol 2014 ; 33 : 1–12. [PubMed] [Google Scholar]
- Asmus F, Salih F, Hjermind LE, et al. Myoclonus-dystonia due to genomic deletions in the epsilon-sarcoglycan gene. Ann Neurol 2005 ; 58 : 792–797. [CrossRef] [PubMed] [Google Scholar]
- Ghaoui R, Cooper ST, Lek M, et al. Use of whole-exome sequencing for diagnosis of limb-girdle muscular dystrophy: outcomes and lessons learned. JAMA Neurol 2015 ; 72 : 1424–1432. [Google Scholar]
- Liu W, Pajusalu S, Lake NJ, et al. Estimating prevalence for limb-girdle muscular dystrophy based on public sequencing databases. Genet Med 2019 ; 21 : 2512–2520. [CrossRef] [PubMed] [Google Scholar]
- Trabelsi M, Kavian N, Daoud F, et al. Revised spectrum of mutations in sarcoglycanopathies. Eur J Hum Genet 2008 ; 16 : 793–803. [Google Scholar]
- Dalichaouche I, Sifi Y, Roudaut C, et al. γ-sarcoglycan and dystrophin mutation spectrum in an Algerian cohort. Muscle and Nerve 2017 ; 56 : 129–135. [CrossRef] [Google Scholar]
- Alavi A, Esmaeili S, Nilipour Y, et al. LGMD2E is the most common type of sarcoglycanopathies in the Iranian population. J Neurogenet 2017 ; 31 : 161–169. [PubMed] [Google Scholar]
- Moore SA, Shilling CJ, Westra S, et al. Limb-girdle muscular dystrophy in the United States. J Neuropathol Exp Neurol 2006 ; 65 : 995–1003. [CrossRef] [PubMed] [Google Scholar]
- Alonso-Pérez J, González-Quereda L, Bello L, et al. New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy. Brain 2020. doi:10.1093/brain/awaa228. [Google Scholar]
- Passos-Bueno MR, Vainzof M, Moreira ES, Zatz M. Seven autosomal recessive limb-girdle muscular dystrophies in the Brazilian population: from LGMD2A to LGMD2G. Am J Med Genet 1999 ; 82 : 392–398. [Google Scholar]
- Piccolo F, Jeanpierre M, Leturcq F, et al. A founder mutation in the gamma-sarcoglycan gene of gypsies possibly predating their migration out of India. Hum Mol Genet 1996 ; 5 : 2019–2022. [CrossRef] [PubMed] [Google Scholar]
- Roberds SL, Leturcq F, Allamand V, et al. Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy. Cell 1994 ; 78 : 625–633. [CrossRef] [PubMed] [Google Scholar]
- Eymard B, Romero NB, Leturcq F, et al. Primary adhalinopathy (α-sarcoglycanopathy): clinical, pathologic, and genetic correlation in 20 patients with autosomal recessive muscular dystrophy. Neurology 1997 ; 48 : 1227–1234. [Google Scholar]
- Semplicini C, Vissing J, Dahlqvist JR, et al. Clinical and genetic spectrum in limb-girdle muscular dystrophy type 2E. Neurology 2015 ; 84 : 1772–1781. [Google Scholar]
- Xie Z, Hou Y, Yu M, et al. Clinical and genetic spectrum of sarcoglycanopathies in a large cohort of Chinese patients. Orphanet J Rare Dis 2019 ; 14 : 43. [CrossRef] [PubMed] [Google Scholar]
- Tasca G, Monforte M, Díaz-Manera J, et al. MRI in sarcoglycanopathies: a large international cohort study. J Neurol Neurosurg Psychiatry 2018 ; 89 : 72–77. [CrossRef] [PubMed] [Google Scholar]
- Schade van Westrum SM, Dekker LRC, de Voogt WG, et al. Cardiac involvement in Dutch patients with sarcoglycanopathy: a cross-sectional cohort and follow-up study. Muscle Nerve 2014 ; 50 : 909–913. [Google Scholar]
- Kyriakides T, Angelini C, Vilchez J, Hilton-Jones D. European federation of the neurological societies guidelines on the diagnostic approach to paucisymptomatic or asymptomatic hyperCKemia. Muscle Nerve 2020; 61 : E14–5. [Google Scholar]
- Fanin M, Melacini P, Boito C, Pegoraro E, Angelini C. LGMD2E patients risk developing dilated cardiomyopathy. Neuromuscul Disord 2003 ; 13 : 303–309. [CrossRef] [PubMed] [Google Scholar]
- Politano L, Nigro V, Passamano L, et al. Evaluation of cardiac and respiratory involvement in sarcoglycanopathies. Neuromuscul Disord 2001 ; 11 : 178–185. [CrossRef] [PubMed] [Google Scholar]
- Sveen ML, Thune JJ, Køber L, Vissing J. Cardiac involvement in patients with limb-girdle muscular dystrophy type 2 and becker muscular dystrophy. Arch Neurol 2008 ; 65 : 1196–1201. [CrossRef] [PubMed] [Google Scholar]
- Guimarães-Costa R, Fernández-Eulate G, Wahbi K, et al. Clinical correlations and long-term follow-up in 100 patients with sarcoglycanopathies. Eur J Neurol 2020; Oct 14. doi:10.1111/ene.14592. [Google Scholar]
- Boito C, Fanin M, Siciliano G, Angelini C, Pegoraro E. Novel sarcoglycan gene mutations in a large cohort of Italian patients. J Med Genet 2003 ; 40 : e67. [CrossRef] [PubMed] [Google Scholar]
- Merlini L, Kaplan JC, Navarro C, et al. Homogeneous phenotype of the gypsy limb-girdle MD with the γ- sarcoglycan C283Y mutation. Neurology 2000 ; 54 : 1075–1079. [Google Scholar]
- Guglieri M, Magri F, D’Angelo MG, et al. Clinical, molecular, and protein correlations in a large sample of genetically diagnosed Italian limb girdle muscular dystrophy patients. Hum Mutat 2008 ; 29 : 258–266. [CrossRef] [PubMed] [Google Scholar]
- Winckler PB, da Silva AMS, Coimbra-Neto AR, et al. Clinicogenetic lessons from 370 patients with autosomal recessive limb-girdle muscular dystrophy. Clin Genet 2019 ; 96 : 341–353. [CrossRef] [PubMed] [Google Scholar]
- Sandonà D, Betto R. Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects. Expert Rev Mol Med 2009 ; 11 : e28. [CrossRef] [PubMed] [Google Scholar]
- Bianchini E, Fanin M, Mamchaoui K, Betto R, Sandonà D. Unveiling the degradative route of the V247M a-sarcoglycan mutant responsible for LGMD-2D. Hum Mol Genet 2014 ; 23 : 3746–3758. [CrossRef] [PubMed] [Google Scholar]
- Poupiot J, Costa Verdera H, Hardet R, et al. Role of rregulatory T cell and effector T cell exhaustion in liver-mediated transgene tolerance in muscle. Mol Ther Methods Clin Dev 2019 ; 15 : 83–100. [CrossRef] [PubMed] [Google Scholar]
- Israeli D, Cosette J, Corre G, et al. An AAV-SGCG dose-response study in a γ-sarcoglycanopathy mouse model in the context of mechanical stress. Mol Ther Methods Clin Dev 2019 ; 13 : 494–502. [CrossRef] [PubMed] [Google Scholar]
- Zhu T, Zhou L, Mori S, et al. Sustained whole-body functional rescue in congestive heart failure and muscular dystrophy hamsters by systemic gene transfer. Circulation 2005 ; 112 : 2650–2659. [CrossRef] [PubMed] [Google Scholar]
- Li J, Wang D, Qian S, Chen Z, Zhu T, Xiao X. Efficient and long-term intracardiac gene transfer in δ-sarcoglycan-deficiency hamster by adeno-associated virus-2 vectors. Gene Ther 2003 ; 10 : 1807–1813. [CrossRef] [PubMed] [Google Scholar]
- Xiao X, Li J, Tsao Y-P, Dressman D, Hoffman EP, Watchko JF. Full functional rescue of a complete muscle (TA) in dystrophic hamsters by adeno-associated virus vector-directed gene therapy. J Virol 2000 ; 74 : 1436–1442. [CrossRef] [PubMed] [Google Scholar]
- Cordier L, Hack AA, Scott MO, et al. Rescue of skeletal muscles of γ-sarcoglycan- deficient mice with adeno-associated virus-mediated gene transfer. Mol Ther 2000 ; 1 : 119–129. [CrossRef] [PubMed] [Google Scholar]
- Cordier L, Gao GP, Hack AA, et al. Muscle-specific promoters may be necessary for adeno-associated virus-mediated gene transfer in the treatment of muscular dystrophies. Hum Gene Ther 2001 ; 12 : 205–215. [Google Scholar]
- Li J, Dressman D, Tsao YP, et al. rAAV vector-mediated sarcogylcan gene transfer in a hamster model for limb girdle muscular dystrophy. Gene Ther 1999 ; 6 : 74–82. [CrossRef] [PubMed] [Google Scholar]
- Mendell JR, Rodino-Klapac LR, Rosales XQ, et al. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Ann Neurol 2010 ; 68 : 629–638. [CrossRef] [PubMed] [Google Scholar]
- Rodino-Klapac LR, Lee JS, Mulligan RC, et al. Lack of toxicity of alpha-sarcoglycan overexpression supports clinical gene transfer trial in LGMD2D. Neurology 2008 ; 71 : 240–247. [Google Scholar]
- Pacak CA, Walter GA, Gaidosh G, et al. Long-term skeletal muscle protection after gene transfer in a mouse model of LGMD-2D. Mol Ther 2007 ; 15 : 1775–1781. [CrossRef] [PubMed] [Google Scholar]
- Fougerousse F, Bartoli M, Poupiot J, et al. Phenotypic correction of α-sarcoglycan deficiency by intra-arterial injection of a muscle-specific serotype 1 rAAV vector. Mol Ther 2007 ; 15 : 53–61. [Google Scholar]
- Dressman D, Araishi K, Imamura M, et al. Delivery of α- and β-sarcoglycan by recombinant adeno-associated virus: efficient rescue of muscle, but differential toxicity. Hum Gene Ther 2002 ; 13 : 1631–1646. [Google Scholar]
- Pozsgai ER, Griffin DA, Heller KN, Mendell JR, Rodino-Klapac LR. Systemic AAV-mediated β-sarcoglycan delivery targeting cardiac and skeletal muscle ameliorates histological and functional deficits in LGMD2E mice. Mol Ther 2017 ; 25 : 855–869. [CrossRef] [PubMed] [Google Scholar]
- Vitiello C, Faraso S, Sorrentino NC, et al. Disease rescue and increased lifespan in a model of cardiomyopathy and muscular dystrophy by combined AAV treatments. PLoS One 2009 ; 4 : e5051. [Google Scholar]
- Goehringer C, Rutschow D, Bauer R, et al. Prevention of cardiomyopathy in δ-sarcoglycan knockout mice after systemic transfer of targeted adeno-associated viral vectors. Cardiovasc Res 2009 ; 82 : 404–410. [CrossRef] [PubMed] [Google Scholar]
- Mendell JR, Rodino-Klapac LR, Rosales-Quintero X, et al. Limb-girdle muscular dystrophy type 2D gene therapy restores α-sarcoglycan and associated proteins. Ann Neurol 2009 ; 66 : 290–297. [CrossRef] [PubMed] [Google Scholar]
- Herson S, Hentati F, Rigolet A, et al. A phase I trial of adeno-associated virus serotype 1-γ-sarcoglycan gene therapy for limb girdle muscular dystrophy type 2C. Brain 2012 ; 135 : 483–492. [CrossRef] [PubMed] [Google Scholar]
- Mendell JR, Chicoine LG, Al-Zaidy SA, et al. Gene delivery for limb-girdle muscular dystrophy type 2d by isolated limb infusion. Hum Gene Ther 2019 ; 30 : 794–801. [Google Scholar]
Liste des tableaux
Caractéristiques épidémiologiques et cliniques des sarcoglycanopathies (d’après le site web neuromuscular.wustl.edu et [11, 24]).
Liste des figures
|
Figure 1. Les quatre sarcoglycanes (α, β, γ, δ) font partie du complexe DGC associé à la dystrophine. © AFM-Téléthon. |
| Dans le texte | |
|
Figure 2. Cas de sarcoglycanopathie-alpha chez un jeune homme de 25 ans présentant une dystrophie musculaire des ceintures évoluant lentement depuis l’enfance. On notera la pseudo-hypertrophie des mollets (A, B), l’hyperlordose (A), les rétractions achilléennes (A, B), le décollement des omoplates (B) et la discrète macroglossie (C). Deux mutations hétérozygotes ont été identifiées dans le gène SGCA (une mutation d’épissage dans l’intron 6 et une mutation dans l’exon 3, la p.R77C). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.