")
")
| Issue |
Med Sci (Paris)
Volume 36, Décembre 2020
Les Cahiers de Myologie
|
|
|---|---|---|
| Page(s) | 28 - 33 | |
| Section | Dystrophies musculaires des ceintures (LGMD) | |
| DOI | https://doi.org/10.1051/medsci/2020239 | |
| Published online | 11 January 2021 | |
La dystrophie musculaire des ceintures de type R9 liée au gène FKRP
État des lieux et perspectives thérapeutiques
Limb-Girdle Muscular Dystrophy type R9 linked to the FKRP gene: state of the art and therapeutic perspectives
1
Centre de Référence des maladies neuromusculaires Nord/Est/Île-de-France, APHP, Groupe Hospitalier Pitié-Salpêtrière, Sorbonne Université, Paris, France.
2
Généthon, 91000 Évry, France.
3
Université Paris-Saclay, Université d’Évry, Inserm, Généthon, unité de recherche Integrare, UMR_S951, 91000 Évry, France
4
Département de Génétique, APHP-Hôpital Bichat Claude Bernard, Paris, France.
* This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les mutations du gène FKRP codant la fukutin-related protein (FKRP) sont à l’origine d’un large éventail de myopathies allant de formes sévères de dystrophies musculaires congénitales associées à des anomalies structurales du système nerveux central, jusqu’à des tableaux de myalgies à l’effort ou d’hyperCKémie asymptomatique, en passant par une forme de dystrophie musculaire des ceintures, la LGMD-R9 (ex-LGMD-2I), pour limb girdle muscular dystrophy récessive de type R9. La LGMD-R9 se caractérise par un déficit proximal des ceintures prédominant initialement aux membres inférieurs, avec une atteinte respiratoire et cardiaque pouvant conditionner le pronostic vital. Le taux sérique de CPK est nettement élevé et s’accompagne, sur la biopsie musculaire, d’une formule dystrophique associée à une réduction de la glycosylation de l’α-dystroglycane visible en immunomarquage et par immunoblot. L’IRM musculaire montre typiquement une atteinte des muscles proximaux (iliopsoas, adducteurs, grands fessiers, quadriceps) avec une relative préservation des muscles de la loge antérieure des cuisses (gracilis et sartorius). L’analyse génétique, par séquençage spécifique du gène FKRP ou d’un panel regroupant l’ensemble des gènes impliqués dans la glycosylation de l’α-dystroglycane, ou bien d’un panel plus large de gènes, confirme généralement le diagnostic, la mutation la plus fréquente étant le faux-sens p.(Leu276Ile). Actuellement, le traitement de la LGMD-R9 est symptomatique, requérant une approche pluridisciplinaire. Une étude prospective d’histoire naturelle de la maladie est en cours en Europe (GNT-015-FKRP). Des approches thérapeutiques inédites sont envisagées, telles que la thérapie génique médiée par des vecteurs dérivés du virus adéno-associé (AAV). Celle-ci est efficace dans les modèles animaux, permettant une correction des défauts de glycosylation de l’a-dystroglycane et une augmentation de sa capacité de liaison à la matrice extracellulaire. En parallèle, des études précliniques ont montré, dans un modèle animal, l’efficacité du ribitol, un pentose alcool retrouvé dans des composés naturels, ce qui a conduit à un essai de phase I dont le développement clinique est en cours.
Abstract
Mutations in the FKRP gene encoding the fukutin-related protein (FKRP) cause a wide spectrum of myopathies, ranging from severe forms of congenital muscular dystrophies associated with structural abnormalities of the central nervous system, to exertional myalgia or asymptomatic hyperCKemia, and to a form of limb girdle muscular dystrophy, LGMD-R9, (ex-LGMD-2I). LGMD-R9 is characterized by a proximal girdle deficit predominantly in the lower limbs to start with, with respiratory and cardiac damage that may affect the vital prognosis. Serum CK levels are markedly elevated and, on muscle biopsy, is detected a dystrophic formula associated with a reduction in the glycosylation of α-dystroglycan by immunostains and immunoblotting. Muscle MRI typically shows damage to proximal muscles (iliopsoas, adductors, gluteus maximus, quadriceps) with relative preservation of the muscles of the anterior compartment of the thighs (gracilis and sartorius). Genetic analysis, by specific sequencing of the FKRP gene or of a panel grouping together all the genes involved in the glycosylation of α-dystroglycan, or a larger panel of genes, generally confirms the diagnosis, the most frequent mutation being the missense p.(Leu276Ile). Currently, treatment of LGMD-R9 is symptomatic, requiring a multidisciplinary approach. A prospective study of the natural history of the disease is currently underway in Europe (GNT-015-FKRP). New therapeutic approaches are envisaged, such as gene therapy mediated by vectors derived from the adeno-associated virus (AAV). This is effective in animal models, allowing correction of defects in the glycosylation of alpha-dystroglycan and an increase in its binding capacity to the extracellular matrix. At the same time, preclinical studies have shown, in an animal model, the efficacy of ribitol, an alcohol pentose found in natural compounds, which has led to a phase I trial whose clinical development is underway.
© 2020 médecine/sciences – Inserm
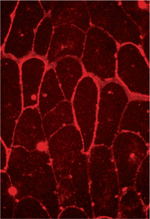
© Theresinha Evangelista
La dystrophie musculaire des ceintures de type R9 (ex-LGMD-2I) est une maladie autosomique récessive consécutive à des mutations du gène FKRP. Ce gène est localisé sur le chromosome 19 (19q13.3) et code la fukutin-related protein (FKRP) [1-3]. La FKRP est une protéine ubiquitaire, plus abondante dans les muscles squelettique et cardiaque. Elle est localisée dans l’appareil de Golgi, dans le réticulum sarcoplasmique et à proximité du sarcolemme (Figure 1). La FKRP a une activité glycosyltransférase et fait partie des dix-neuf enzymes impliquées dans la glycosylation de l’α-dystroglycane. La glycosylation est un processus clé pour maintenir l’intégrité structurale de l’ancrage entre le sarcolemme et la matrice extracellulaire [4].
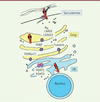 |
Figure 1. Représentation schématique de la localisation cellulaire des protéines impliquées dans la glycosylation de l’a-dystroglycane (adapté de [28]). |
La FKRP a été identifiée grâce à son homologie avec la fukutine, une protéine déjà connue pour son implication dans une dystrophie musculaire congénitale autosomique récessive décrite initialement au Japon (Fukuyama-type congenital muscular dystrophy, FCMD) et caractérisée par l’association d’un phénotype musculaire sévère et de malformations cérébrales (micropolygyrie) [5]. Par la suite, des mutations du gène de la fukutine (FKTN) ont été impliquées dans un spectre clinique plus étendu avec des formes plus sévères mais aussi des formes qui l’étaient moins, notamment sans atteinte du système nerveux central [6].
Des mutations du gène FKRP ont été décrites chez des patients présentant une dystrophie musculaire congénitale (de type MDC1C selon la nomenclature internationale) caractérisée par un début dans les premiers mois de vie, une hypotonie et une faiblesse musculaire, une élévation du taux sérique de CPK, un retard du développement moteur avec retard ou absence d’acquisition de la marche, mais sans atteinte du système nerveux central [7]. Une réduction de la glycosylation de l’α-dystroglycane a été détectée en immunomarquage sur biopsie musculaire et en western blot. Peu après, des mutations du gène FKRP furent associées à une forme de dystrophie musculaire des ceintures (LGMD-R9) associée à une atteinte cardiaque mais sans atteinte structurelle au niveau cérébral [1]. Dans cette forme, la glycosylation de l’α-dystroglycane était moins réduite que dans les cas de MDC1C, ce qui concorde avec une expression phénotypique musculaire plus bénigne. Cette forme de LGMD s’est avérée être une des formes les plus fréquentes de LGMD en Europe, du fait d’une mutation fondatrice (c.826C >A ; p.(Leu276Ile)) dont la fréquence allélique est de ~1/400. Cette mutation est particulièrement fréquente en Europe du nord où la prévalence de la LGMD-R9 se situerait autour de 1/54 000 d’après une étude conduite au Danemark et en Norvège [8].
Des mutations du gène FKRP ont été également détectées chez des patients présentant un tableau de dystrophie musculaire congénitale associée à un retard mental et à des anomalies structurales du système nerveux central telles que des dysplasies du cortex cérébral et/ou des kystes cérébelleux [9], ainsi que chez des patients ayant des anomalies corticales plus sévères encore et des malformations oculaires comparables à celles décrites dans les syndromes de Walker Warburg (WWS) et du « Muscle-Eye-Brain disease » (MEB) [10].
Présentation clinique de la dystrophie musculaire des ceintures de type R9
L’éventail clinique de la myopathie des ceintures liée à des mutations du gène FKRP est très étendu, allant de formes sévères évoquant une dystrophie musculaire de Duchenne (Duchenne-like), avec une progression rapide du déficit moteur et perte de la marche à l’adolescence, jusqu’à des formes plus bénignes avec conservation de la marche, et y compris des tableaux d’hyperCKémie asymptomatique [2, 11]. Il existerait aussi un certain degré de variabilité intrafamiliale.
L’âge des premiers symptômes s’étend de la petite enfance jusqu’à l’âge adulte. Le plus souvent, toutefois, la maladie débute dans la deuxième décennie avec des difficultés liées à une faiblesse musculaire proximale, notamment aux membres inférieurs, avec une démarche dandinante et des difficultés pour la montée des escaliers. Dans une étude compilant les données de trente-huit patients, un tiers d’entre eux rapportaient des symptômes apparus dans l’enfance sous la forme d’une marche digitigrade ou de difficultés pour courir et/ou faire du sport [2].
Dans deux tiers des cas, les patients présentent des crampes et des myalgies à l’effort, notamment aux mollets, ainsi que des épisodes de myoglobinurie dans un tiers des cas [12,13]. Par ailleurs, lors d’épisodes fébriles, les patients peuvent présenter une aggravation brutale, mais réversible, de la faiblesse motrice sur une durée pouvant varier de quelques jours à plus d’un mois. Lors de ces épisodes, l’augmentation du taux des CPK peut être très importante [13].
Typiquement, le phénotype clinique de la LGMD-R9 se caractérise par une faiblesse proximale des ceintures avec une relative préservation de la force distale (Figure 2). L’atteinte prédomine initialement aux membres inférieurs, avec une atteinte des psoas, des adducteurs de hanche et des quadriceps. Aux membres supérieurs, on observe souvent un décollement des omoplates. Le déficit prédomine sur les muscles de la ceinture scapulaire ainsi que sur les triceps et les biceps brachiaux [2, 14, 15]. Les formes plus sévères avec un tableau Duchenne-like peuvent avoir aussi une atteinte prédominant initialement aux membres supérieurs [14]. L’âge de début dans ces formes est plus précoce avec une perte de la marche notée au cours des premières décennies de la vie (Figure 3).
 |
Figure 2. Phénotype typique de LGMD-R9. On observe un décollement des omoplates (A), ainsi qu’une hypertrophie des mollets (B) chez une patiente encore capable de marcher malgré un déficit musculaire modéré à prédominance proximale. |
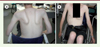 |
Figure 3. Phénotype Duchenne-like. On note le décollement des omoplates (A) ainsi qu’une amyotrophie des membres supérieurs à prédominance proximale. Le déficit moteur a conduit à une perte de la marche (B). |
Les patients présentent classiquement une hypertrophie des mollets et parfois de la langue [2, 14]. En général, les rétractions musculo-tendineuses ne sont ni fréquentes ni au premier plan, mais certains patients avec un phénotype plus sévère peuvent avoir des rétractions marquées, notamment aux membres inférieurs. La présence d’une lordose lombaire est possible, tandis que la scoliose et/ou la rigidité spinale sont rares.
L’atteinte respiratoire est présente chez environ 50 % des patients, et peut survenir alors qu’ils sont encore en capacité de marcher. Une ventilation nocturne non-invasive est nécessaire dans 25 à 50 % des patients [2, 11]. L’atteinte diaphragmatique est précoce avec une diminution de la capacité vitale (CVF) en décubitus de plus de 10 % par rapport à la mesure en position assise. Il est souhaitable d’examiner la CVF dans les deux positions lors des épreuves fonctionnelles respiratoires.
L’atteinte cardiaque fait aussi partie de la maladie. Elle est présente chez 30 à 55 % des patients selon les séries [11, 12] et conditionne largement le pronostic vital. Elle se traduit le plus souvent par la présence d’une cardiomyopathie dilatée. L’échographie cardiaque montre habituellement des défauts de contractilité ainsi qu’une diminution de la fraction d’éjection du ventricule gauche (FEVG). Récemment, une étude longitudinale évaluant la fonction cardiaque chez ces patients a montré une diminution de 0 à 4 % par an de la FEVG [17].
L’IRM cardiaque montre souvent une fibrose ainsi qu’une dégénérescence graisseuse du myocarde associées à la diminution de la fonction contractile et du volume du ventricule gauche [18]. Cet examen apparaît plus sensible pour détecter des anomalies cardiaques comparativement aux explorations conventionnelles (ECG et échocardiogramme). Il n’existe pas de corrélation entre l’atteinte cardiaque et la gravité du déficit musculaire [12, 18]. Par conséquent, la surveillance cardiaque doit être systématique et régulière chez ces patients.
Diagnostic et explorations complémentaires
Le diagnostic repose sur la combinaison des signes cliniques, de la biopsie musculaire, de l’imagerie musculaire et de l’analyse génétique.
Le taux sérique de CPK est élevé et peut atteindre jusqu’à dix fois la limite supérieure de la normale. La biopsie musculaire (Figure 4) montre des lésions compatibles avec un processus dystrophique telles que des fibres arrondies, de taille inégale, ou des fibres en nécrose et en régénération. En immunomarquage, on observe une réduction de la glycosylation de l’α-dystroglycane, confirmée par immunoblot. En revanche, il n’existe pas de corrélation stricte entre la sévérité de l’atteinte clinique et la réduction du signal de la FKRP en immunomarquage ou en immunoblot. On peut également objectiver une réduction secondaire du marquage des chaines α2 et β1 de la laminine-211, alors que le marquage de la dystrophine est généralement normal par ailleurs.
 |
Figure 4. Biopsie musculaire. On observe un aspect dystrophique des fibres musculaires caractérisé par une variation de taille des fibres et une augmentation du tissu conjonctif en H&E (A). Réduction de l’immunomarquage de l’α-dystroglycane (B) alors que celui du β-dystroglycane est normal. |
L’IRM musculaire des membres inférieurs montre typiquement des anomalies de signal, une atrophie et une infiltration graisseuse des muscles proximaux tels que l’iliopsoas, les adducteurs et les grands fessiers, ainsi que le vaste médial du quadriceps avec une relative conservation des muscles de la loge antérieure des cuisses, du gracilis et du sartorius [19].
L’analyse génétique confirme le diagnostic, soit directement par séquençage du gène FKRP par la méthode Sanger, soit le plus souvent aujourd’hui, par séquençage à haut débit de type NGS (Next Generation Sequencing) d’un panel de gènes connus (19 à ce jour) pouvant être impliqués dans la glycosylation de l’α-dystroglycane : B3GALNT2, B3GNT1, DAG1, DOLK, DPM1, DPM2, DPM3, FKRP, FKTN, GMPPB, ISPD (CRPPA), LARGE, POMGNT1, POMGNT2 (GTDC2), POMK, POMT1, POMT2, TMEM5=RXYLT1, INPP5K (Figure 5).
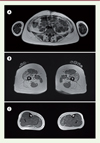 |
Figure 5. IRM musculaire. On observe une atrophie graisseuse des muscles paravertébraux (A). Au niveau des cuisses, il existe une atteinte prédominante sur les muscles de la loge postérieure des cuisses avec préservation des muscles gracilis et sartorius (B). Aux jambes, on note une atrophie graisseuse modérée des muscles gastrocnémiens (C). |
Comme évoqué précédemment, le défaut génétique le plus fréquemment rencontré est la mutation c.826C>A qui conduit à un changement d’acide aminé (isoleucine à la place de leucine) en position 276. Les patients homozygotes pour ce variant présentent typiquement un phénotype de dystrophie des ceintures (LGMD) ou des formes encore moins sévères (hyperCKémie chronique). Ce variant n’est jamais associé à des formes congénitales graves, qu’il s’agisse d’un syndrome de WWS ou d’un syndrome MEB. En revanche, les patients porteurs de ce variant à l’état hétérozygote, en association avec un autre variant pathologique du même gène, peuvent présenter un phénotype plus sévère, de type Duchenne-like, avec un début dans la première décennie, une perte de la marche à l’adolescence voire plus tôt, et la nécessité d’une assistance ventilatoire précoce.
Perspectives thérapeutiques
Il n’existe actuellement aucun traitement spécifique de cette forme de dystrophie musculaire des ceintures. Le traitement symptomatique est basé sur une approche pluri-disciplinaire impliquant, notamment, une surveillance cardiaque et respiratoire régulière. L’atteinte cardiaque conditionnant souvent le pronostic vital, il est impératif de mettre en place, le moment venu, un traitement pharmacologique au long cours comportant au minimum un inhibiteur de l’enzyme de conversion de l’angiotensine, à l’instar de ce qui est recommandé pour la prise en charge de la cardiomyopathie de la dystrophie musculaire de Duchenne. La détection précoce d’une insuffisance respiratoire est capitale et peut conduire à la mise en place d’une ventilation non-invasive. La prise en charge rééducative enfin, peut améliorer les capacités fonctionnelles résiduelles des patients.
La LGMD-R9 est une affection autosomique récessive. Il en découle une perte de fonction de la protéine FKRP. Une approche thérapeutique par remplacement de gène est donc adaptée dans ce cas précis. A ce jour, cette stratégie pour les maladies neuromusculaires repose sur un transfert de gène médié par des vecteurs dérivés du virus AAV. Malgré les défis et les revers historiques qu’elle a connus, la thérapie génique médiée par l’AAV reste l’approche la plus prometteuse pour l’administration des gènes de pathologies neuromusculaires et ce pour plusieurs raisons : absence de pathogénicité et d’intégration génomique de l’AAV, faible réponse immunitaire induite comparée à d’autres vecteurs, capacité élevée à transduire des cellules musculaires, qu’elles soient en division ou non. Il existe des exemples récents de développement clinique réussi ayant conduit à des autorisations de mise sur le marché aux États-Unis et/ou en Europe, comme le voretigène néparvovec (Luxturna®) et l’onasemnogene abeparvovec-xioi (Zolgensma®). Plusieurs études cliniques destinées à évaluer l’administration systémique de virus AAV sont également en cours chez des patients atteints d’autres maladies neuromusculaires telles que la dystrophie musculaire de Duchenne (NCT04240314, NCT03362502, NCT03368742, NCT03375164), la LGMD-R4 (NCT03652259), la myopathie myotubulaire liée à l’X (NCT03199469) et l’amyotrophie spinale infantile (NCT03461289, NCT03505099, NCT03837184).
La preuve de principe de l’efficacité du transfert du gène FKRP médié par un AAV afin de restaurer le phénotype de la souris déficiente en FKRP a été rapportée dans plusieurs études dont celle de l’équipe de Généthon [20-24]. Après avoir généré un modèle animal portant la mutation c.826C>A, cette dernière a administré par voie systémique un vecteur AAV portant la séquence codante du gène FKRP sous la dépendance transcriptionnelle d’un promoteur musculaire. Les résultats ont abouti à une expression du transgène dose-dépendante tant au niveau de l’ARN messager qu’au niveau protéique. Une correction des défauts de glycosylation de l’a-dystroglycane, et en conséquence une augmentation de sa capacité de liaison à la laminine-211, ont pu être démontrées. Ces effets moléculaires ont permis une correction de la pathologie observée sur coupes histologiques de muscle ainsi qu’une restauration de la force [22]. Dans l’ensemble, ces données constituent des arguments en faveur de l’utilisation du transfert du gène FKRP médié par l’AAV comme approche thérapeutique de la LGMD-R9. Le laboratoire Généthon se prépare activement dans la perspective d’un essai clinique à court/moyen terme. Le chemin vers un tel essai reste toutefois complexe : il comprend pour la partie préclinique une étape éventuelle d’amélioration du vecteur, la détermination de la dose minimale efficace par le biais d’études chez l’animal, et des études réglementaires de toxicité et de biodistribution. Pour la partie production, il faut mettre au point des méthodes industrielles respectant les normes GMP (Good Manufacturing Practices), une procédure de contrôle-qualité et une production des lots cliniques qui se fait dans des unités dédiées. Pour ce qui concerne le développement clinique, la préparation et soumission du dossier aux agences est en cours. Grâce à une collaboration internationale, une étude prospective d’histoire naturelle est en cours d’achèvement en Europe (GNT-015-FKRP). Elle implique des centres danois, allemands, anglais et français. En France, l’étude est coordonnée par le Centre de référence des maladies neuromusculaires Nord/Est/Île-de-France à l’Institut de myologie (Paris). Le but de cette étude est de caractériser l’évolution et l’histoire naturelle de la LGMD-R9 ainsi que d’identifier des marqueurs cliniques, fonctionnels et d’imagerie qui pourraient garantir le succès à venir des essais thérapeutiques.
En parallèle aux approches de thérapie génique, des études précliniques suggèrent que le ribitol, un pentose alcool retrouvé dans des composés naturels, pourrait avoir des vertus thérapeutiques. La FKRP est une enzyme participant à la biosynthèse de la chaîne glycanique de l’α-DG en catalysant l’addition de ribitol-5-phosphate sur la chaîne du glycan O-mannosyl précédemment formée sous l’action des enzymes POMT1/POMT2, POMGNT2, B3GALNT2 et POMK [25]. Le substrat de cette réaction est le cytidine di-phosphate, (CDP)-ribitol, qui est synthétisé par la protéine isoprenoid synthase (ISPD) [26]. Il a été montré que l’administration orale de ribitol conduisait à une augmentation de synthèse de CDP-ribitol et restaurait à un niveau thérapeutique la glycosylation de l’α−dystroglycane dans un modèle murin de FKRP portant une mutation faux-sens [27]. Le mécanisme sous-tendant cet effet positif est de compenser par un excès de substrat la diminution de la fonction de la FKRP mutée, la condition étant de facto que les mutations n’aient pas aboli entièrement la fonction enzymatique ou la possibilité d’attachement du substrat à la chaîne glycanique. En prélude à un passage chez des patients atteints de LGMD-R9, un essai de phase I sur volontaires sains, sponsorisé par ML Bio Solutions, a débuté cette année pour évaluer la sécurité et la dose optimale après administration orale de ce produit.
En conclusion, l’ensemble de ces travaux permet d’espérer que dans un futur proche des solutions thérapeutiques pourront être mises à disposition des patients présentant une LGMD-R9.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Brockington M, Yuva Y, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 21 as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet 2001 ; 10 : 2851–2859. [CrossRef] [PubMed] [Google Scholar]
- Poppe M, Cree L, Bourke J, et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology 2003 ; 60 : 1246–1251. [Google Scholar]
- Hara Y, Balci-Hayta B, Yoshida-Moriguchi T, et al. A Dystroglycan mutation associated with limb-girdle muscular dystrophy. N Engl J Med 2011 ; 34464 : 939–946. [Google Scholar]
- Alhamidi M, Kjeldsen Buvang E, et al. Fukutin-related protein resides in the Golgi cisternae of skeletal muscle fibres and forms disulfide-linked homodimers via an N-terminal interaction. PLoS One 2011 ; 6 : [Google Scholar]
- Kobayashi K, Nakahori Y, Miyake M, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 1998 ; 394 : 388–392. [CrossRef] [PubMed] [Google Scholar]
- Muntoni F, Torelli S, Wells DJ, et al. Muscular dystrophies due to glycosylation defects. Curr Opin Neurol 2011 ; 24 : 437–442. [CrossRef] [PubMed] [Google Scholar]
- Brockington M, Blake DJ, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin α2 deficiency and abnormal glycosylation of α-dystroglycan. Am J Hum Genet 2001 ; 69 : 1198–1209. [Google Scholar]
- Stensland E, Lindal S, Jonsrud C, et al. Prevalence, mutation spectrum and phenotypic variability in Norwegian patients with limb girdle muscular dystrophy 2I. Neuromuscul Disord 2011 ; 21 : 41–46. [CrossRef] [PubMed] [Google Scholar]
- Topaloglu H, Brockington M, Yuva Y, et al. FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology 2003 ; 60 : 988–992. [Google Scholar]
- Beltran-Valero de Bernabé D, Voit T, Longman C, et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet 2004 ; 41 : [Google Scholar]
- Poppe M, Bourke J, Eagle M, et al. Cardiac and respiratory failure in limb-girdle muscular dystrophy 2I. Ann Neurol 2004 ; 56 : 738–741. [CrossRef] [PubMed] [Google Scholar]
- Sveen ML, Schwartz M, Vissing J. High prevalence and phenotype-genotype correlations of limb girdle muscular dystrophy type 2I in Denmark. Ann Neurol 2006 ; 59 : 808–815. [CrossRef] [PubMed] [Google Scholar]
- Carlson CR, McGaughey SD, Eskuri JM, et al. Illness-associated muscle weakness in dystroglycanopathies. Neurology 2017 ; 89 : 2374–2380. [Google Scholar]
- Mercuri E, Brockington M, Straub V, et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann Neurol 2003 ; 53 : 537–542. [CrossRef] [PubMed] [Google Scholar]
- Bourteel H, Vermersch P, Cuisset JM, et al. Clinical and mutational spectrum of limb-girdle muscular dystrophy type 2I in 11 French patients. J Neurol Neurosurg Psychiatry 2009 ; 80 : 1405–08 7. [CrossRef] [PubMed] [Google Scholar]
- Petri H, Sveen ML, Thune JJ, et al. Progression of cardiac involvement in patients with limb-girdle type 2 and Becker muscular dystrophies: a 9-year follow-up study. Int J Cardiol 2015 ; 182 : 403–411. [CrossRef] [PubMed] [Google Scholar]
- Wahbi K, Meune C, Hammouda EH, et al. Cardiac assessment of limb-girdle muscular dystrophy 2I patients: an echography, Holter ECG and magnetic resonance imaging study. Neuromuscul Disord 2008 ; 18 : 650–655. [CrossRef] [PubMed] [Google Scholar]
- Willis TA, Hollingsworth KG, Coombs A, et al. Quantitative magnetic resonance imaging in limb-girdle muscular dystrophy 2i: a multinational cross-sectional study. PLoS One 2014 ; 9 : [Google Scholar]
- Xu L, Lu PJ, Wang CH, et al. Adeno-associated virus 9 mediated FKRP gene therapy restores functional glycosylation of α-dystroglycan and improves muscle functions. Mol Ther 2013 ; 21 : 1832–1840. [CrossRef] [PubMed] [Google Scholar]
- Qiao C, Wang CH, Zhao C, et al. Muscle and heart function restoration in a limb girdle muscular dystrophy 2I (LGMD2I) mouse model by systemic FKRP gene delivery. Mol Ther 2014 ; 22 : 1890–1899. [CrossRef] [PubMed] [Google Scholar]
- Gicquel E, Maizonnier N, Foltz SJ, et al. AAV-mediated transfer of FKRP shows therapeutic efficacy in a murine model but requires control of gene expression. Hum Mol Genet 2017 ; 26 : 1952–1965. [CrossRef] [PubMed] [Google Scholar]
- Vannoy CH, Xiao W, Lu P, et al. Efficacy of gene therapy is dependent on disease progression in dystrophic mice with mutations in the FKRP gene. Mol Ther Methods Clin Dev 2017 ; 5 : 31–42. [CrossRef] [PubMed] [Google Scholar]
- Vannoy CH, Leroy V, Lu QL. Dose-dependent effects of fkrp gene-replacement therapy on functional rescue and longevity in dystrophic mice. Mol Ther Methods Clin Dev 2018 ; 11 : 106–120. [CrossRef] [PubMed] [Google Scholar]
- Kawanaga M, Kobayashi K, Tajiri M, et al. Identification of a post-translational modification with ribitol-phosphate and its defect in muscular dystrophy. Cell Reports 2016 ; 14 : 2209–2223. [CrossRef] [PubMed] [Google Scholar]
- Gerin I, Ury B, Breloy I, et al. ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto α-dystroglycan. Nat Commun 2016; 7. [Google Scholar]
- Cataldi MP, Lu P, Blaeser A, et al. Ribitol restores functionally glycosylated α-dystroglycan and improves muscle function in dystrophic FKRP-mutant mice. Nat Commun 2018; 9. [Google Scholar]
- Barresi R, Campbell KP. Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci. 2006 ; 119 : 199–207. [Google Scholar]
Liste des figures
|
Figure 1. Représentation schématique de la localisation cellulaire des protéines impliquées dans la glycosylation de l’a-dystroglycane (adapté de [28]). |
| Dans le texte | |
|
Figure 2. Phénotype typique de LGMD-R9. On observe un décollement des omoplates (A), ainsi qu’une hypertrophie des mollets (B) chez une patiente encore capable de marcher malgré un déficit musculaire modéré à prédominance proximale. |
| Dans le texte | |
|
Figure 3. Phénotype Duchenne-like. On note le décollement des omoplates (A) ainsi qu’une amyotrophie des membres supérieurs à prédominance proximale. Le déficit moteur a conduit à une perte de la marche (B). |
| Dans le texte | |
|
Figure 4. Biopsie musculaire. On observe un aspect dystrophique des fibres musculaires caractérisé par une variation de taille des fibres et une augmentation du tissu conjonctif en H&E (A). Réduction de l’immunomarquage de l’α-dystroglycane (B) alors que celui du β-dystroglycane est normal. |
| Dans le texte | |
|
Figure 5. IRM musculaire. On observe une atrophie graisseuse des muscles paravertébraux (A). Au niveau des cuisses, il existe une atteinte prédominante sur les muscles de la loge postérieure des cuisses avec préservation des muscles gracilis et sartorius (B). Aux jambes, on note une atrophie graisseuse modérée des muscles gastrocnémiens (C). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.