")
")
| Issue |
Med Sci (Paris)
Volume 40, Number 12, Décembre 2024
Épigénétique : développement et destin cellulaire
|
|
|---|---|---|
| Page(s) | 892 - 903 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2024177 | |
| Published online | 20 December 2024 | |
Reprogrammation, lignée germinale et empreinte génomique parentale
Epigenetic reprogramming, germline and genomic imprinting
IGMM, Univ Montpellier, CNRS, Montpellier, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Le maintien de l’identité cellulaire par l’épigénome est crucial pour le développement d’un individu. Les marques chromatiniennes assurent la stabilité de l’expression génique. La reprogrammation épigénétique est un phénomène naturel au cours du développement des mammifères, en particulier lors de l’émergence de la lignée germinale. Les cellules germinales primordiales (PGC) subissent un profond remodelage de la chromatine, comprenant une déméthylation globale de l’ADN et l’effacement des empreintes parentales. Cette revue introduit les notions de reprogrammation épigénétique, y compris sa découverte et ses étapes principales, la lignée germinale murine qui inclut ses changements transcriptionnels et chromatiniens, ainsi que l’empreinte génomique parentale, ses mécanismes et son importance chez l’homme.
Abstract
The memory of cellular identity is crucial for the correct development of an individual and is maintained throughout life by the epigenome. Chromatin marks, such as DNA methylation and histone modifications, ensure the stability of gene expression programmes over time and through cell division. Loss of these marks can lead to severe pathologies, including cancer and developmental syndromes. However, reprogramming of cellular identity is also a natural phenomenon that occurs early in mammalian development, particularly in the germ line, which enables the production of mature and functional gametes. The germ line transmits genetic and epigenetic information to the next generation, contributing to the survival of the species. Primordial germ cells (PGCs) undergo extensive chromatin remodelling, including global DNA demethylation and erasure of the parental imprints. This review introduces the concept of epigenetic reprogramming, its discovery and key steps, as well as the transcriptional and chromatin changes that accompany germ cell formation in mice. Finally, we discuss the epigenetic mechanisms of genomic imprinting, its discovery, regulation and relevance to human disease.
© 2024 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (© Biorender.com).
L’épigénétique se définit aujourd’hui comme l’en semble des mécanismes qui permettent la régulation de l’expression des gènes, de manière stable et héritable à travers les divisions cellulaires. C’est un processus réversible qui à lieu en absence de changement de la séquence de l’ADN. En 1942, Conrad Waddington a défini pour la première fois le terme d’épigénétique comme l’étude des interactions entre génotype, phénotype et environnement, qui permettent d’expliquer qu’une cellule possédant un génome unique puisse donner naissance à de multiples types cellulaires différents [1]. Les marques épigénétiques les plus connues sont la méthylation de l’ADN et les modifications post-traductionnelles des histones (méthylation et acétylation entre autres), qui conduisent à un remodelage de la chromatine [2]. Ces différents processus consolident le profil d’expression d’une cellule, ce qui lui permet d’accéder à des destins cellulaires distincts et spécialisés. Bien que les cellules diploïdes d’un individu partagent un même génome, elles présentent un vaste éventail d’épigénomes, sous-tendant différentes entités cellulaires. Une fois différenciée, il est important pour la cellule de conserver la mémoire de ce choix, de cette identité cellulaire essentielle pour le développement correct d’un individu. Au cours du développement d’un individu, des moments précis existent au cours desquels ces marques épigénétiques sont effacées. Il s’agit de la reprogrammation épigénétique, durant laquelle la plupart des marques chromatiniennes sont remodelées. En effet, au contraire des mutations de l’ADN, les modifications épigénétiques sont réversibles. Cette revue décrit les principes qui ont conduit au prix Nobel de Médecine ou Physiologie attribué à John Gurdon et Shinya Yamanaka en 2012 pour la découverte de la reprogrammation épigénétique, ainsi que ses grandes caractéristiques et étapes au cours du développement d’un individu [3]. Dans une seconde partie, nous nous consacrerons à l’émergence de la lignée germinale chez les mammifères, qui conduit à la création des deux cellules germinales spécialisées, l’ovule et le spermatozoïde, dont la fonction est indispensable à la perpétuation des espèces. La spécification embryonnaire des cellules germinales constitue un changement de destin cellulaire majeur qui s’accompagne d’une reprogrammation globale du génome. En prenant la souris comme modèle, nous discuterons les changements transcriptionnels et chromatiniens à l’origine de l’émergence de la lignée germinale [4]. Enfin, dans une troisième partie, nous introduirons la notion d’empreinte génomique parentale, qui conduit à l’expression mono-allélique d’un ensemble de gènes, en fonction de leur origine parentale. L’empreinte parentale a été révélée par des expériences d’embryologie dans les années 1980. Sa mécanique fine est depuis étudiée car il s’agit d’un exemple phare de régulation génique par des mécanismes épigénétiques, incluant des ARN non codants et un remodelage chromatinien. Le cycle de l’empreinte (effacement et réapposition dans la lignée germinale, puis maintien et lecture lors du développement de l’individu) permet une meilleure compréhension de la reprogrammation épigénétique au cours du développement [5]. Nous discuterons également de l’importance des gènes soumis à empreinte parentale dans certains syndromes humains, ainsi que de la découverte récente de nouveaux gènes soumis à empreinte dite non canonique [6].
La reprogrammation épigénétique
Définition et concepts de base des marques épigénétiques
La reprogrammation épigénétique se définit par un remodelage global de l’épigénome : méthylation de l’ADN, marques d’histones, empreinte parentale et chez les femelles inactivation du chromosome X, un mécanisme épigénétique permettant la compensation de dose entre les sexes. Cette reprogrammation permet la réinitialisation de l’épigénome d’une cellule différenciée et spécialisée. Dans le cas du clonage, la reprogrammation du noyau d’une cellule somatique permet de retrouver un état de pluripotence et la capacité de se différencier vers les trois feuillets germinaux et, par la suite, vers toutes les cellules d’un nouvel individu. Ainsi, la reprogrammation peut être vue comme le chemin inverse des voies de différenciation. Dans les cellules eucaryotes, le matériel génétique est organisé dans le noyau en territoires chromosomiques (environ 100 à 3 000 mégabases [Mb]), puis en compartiments A/B (environ 10 à 50 Mb), en domaines d’association topologiques (TAD) (< 5 Mb) et enfin en boucles de chromatine (< 500 kilobases [kb]) [7]. Les différentes marques chromatiniennes permettent la régulation de la conformation topologique du génome en trois dimensions et l’accessibilité de la chromatine sous la forme de deux états : l’euchromatine, décondensée, riche en gènes et transcriptionnellement active qui est présente dans le compartiment A ; et l’hétérochromatine, condensée, riche en séquences répétées (télomères, satellites, transposons), majoritairement inactive, inaccessible à la machinerie de transcription et présente dans le compartiment B.
La méthylation de l’ADN est généralement associée à la répression transcriptionnelle [8]. Chez les mammifères, les groupements méthyls (-CH3) affectent uniquement les cytosines, en particulier celles impliquées dans des dinucléotides CpG. Dans le contexte d’une cellule somatique, les îlots CpG (séquences à forte densité de CpG typiquement associées aux séquences promotrices) sont généralement hypométhylés, à l’exception de ceux associés aux gènes soumis à empreinte, aux gènes de la lignée germinale et aux gènes du chromosome X inactif chez les individus femelles. Le reste du génome est fortement méthylé, incluant les séquences répétées, les séquences intergéniques, et le corps des gènes. La méthylation des cytosines est catalysée par les ADN méthyltransférases (DNMT pour DNA methyltransferase), DNMT1, DNMT3A, DNMT3B et DNMT3C (présente uniquement chez les rongeurs), toutes dotées d’activité catalytique, ainsi que leur co-facteur DNMT3L (DNA methyltransferase 3 like), sans activité catalytique propre mais qui permet la stimulation de l’activité de DNMT3A et DNMT3B [8]. Les DNMT3 établissent de novo les profils de méthylation des lignages embryonnaires, somatiques ou germinaux, qui sont ensuite perpétués lors des divisions cellulaires par l’activité de maintenance de DNMT1, aidée par son co-facteur UHRF1 (ubiquitin like with PHD and ring finger domains 1). Le méthylome d’un individu est dynamique au cours de la vie, en particulier lors des phases précoces de développement (Figure 1A) caractérisées par deux vagues de reprogrammation : 1) après la fécondation, dans l’embryon, et 2) en amont de la fécondation, dans la lignée germinale primordiale (Figure 1). L’effacement des groupements méthyles peut se produire de manière passive, par dilution à chaque réplication de l’ADN en l’absence de la machinerie de méthylation, ou de façon active, via l’action des déoxygénases TET (ten-eleven translocase) qui, après plusieurs cycles d’oxydation, permettent le remplacement d’une cytosine méthylée par une cytosine non modifiée.
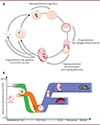 |
Figure 1 Le cycle de programmation et reprogrammation épigénétique au cours du développement murin. A. Les grandes étapes d’apposition et effacement des marques épigénétiques chez la souris. B. Représentation des niveaux globaux de méthylation de l’ADN des gamètes à la souris adulte. TE : trophectoderme ; MCI : masse cellulaire interne ; PE : endoderme primitif ; ExE : ectoderme extra-embryonnaire ; VE : endoderme viscéral ; Epi : épiblaste ; E0,5 : 0,5 jours de développement embryonnaire. |
Les modifications des histones jouent également un rôle prépondérant dans la régulation épigénétique de l’identité cellulaire. Tout comme la méthylation de l’ADN, les modifications des histones sont réversibles et subissent des remaniements drastiques au cours du développement (Figure 1A). Les protéines histones qui forment le nucléosome peuvent être modifiées post-traductionnellement : acétylation, méthylation, phosphorylation, ubiquitination et sumoylation sont les marques les plus étudiées. Plus d’une centaine de marques post-traductionnelles ont été recensées, formant un « code des histones » qui est apposé, lu et effacé par des facteurs chromatiniens (les « writers », « readers » et « erasers ») [9]. Le nombre connu de ces modifications a récemment bondi grâce à l’avènement des techniques de spectrométrie de masse à haute résolution. Les marques H3K4me (méthylation de la lysine 4 de l’histone 3), H3K36me et H3K27ac (acétylation de la lysine 27 de l’histone 3) sont associées à l’euchromatine, alors que les marques H3K27me, H3K9me et H4K20me sont impliquées dans la répression génique. Par ailleurs, H3K27me3 (tri-méthylation de la lysine 27 de l’histone 3) est majoritairement associée à l’hétérochromatine facultative (empreinte parentale non canonique et inactivation du chromosome X) ou à la répression transcriptionnelle dans les régions euchromatiniennes. H3K9me3 marque plus spécifiquement les séquences répétées du génome, telles que les éléments transposables et les séquences d’ADN satellite.
Cycle de (re)programmation lors du développement précoce
Le développement est un processus hautement régulé, qui permet à un zygote totipotent, résultant de la fusion de deux gamètes haploïdes, de former un organisme fonctionnel complet. Ainsi, chaque cycle développemental traduit le passage par des niveaux décroissants de plasticité : de la totipotence à la pluripotence puis à la multipotence, avec finalement l’engagement vers un lignage cellulaire défini, jusqu’à un état de différenciation terminale. Cette restriction progressive de la plasticité est accompagnée de modifications épigénétiques qui garantissent une stabilité et une mémoire des identités cellulaires. La restriction de la plasticité la plus précoce a lieu lors de la divergence entre la masse cellulaire interne (MCI) et le trophectoderme à laquelle sont confrontés les blastomères. La masse cellulaire interne constituera l’embryon proprement dit tandis que le trophectoderme constituera les tissus extra-embryonnaires (Figure 1).
À l’inverse, l’embryon précoce et les cellules germinales primordiales (PGC) sont reprogrammés, avec un effacement global de la méthylation de l’ADN et un remaniement drastique des marques d’histones (Figure 1B et Figure 2). La reprogrammation épigénétique serait essentielle à la fonctionnalité des gamètes en amont de la fécondation, et à l’acquisition de la pluripotence de l’embryon après fécondation. Les gamètes sont des cellules hautement spécialisées avec un fort dimorphisme sexuel. À la fécondation, le génome du spermatozoïde arrive dans l’ovocyte dans un état inactif et extrêmement compacté par des protéines alternatives aux histones, appelées protamines. L’ovocyte, en plus de l’ADN maternel, contient tout le bagage moléculaire permettant d’initier le programme développemental avant l’activation du génome embryonnaire. Il existe donc une importante asymétrie épigénétique entre les génomes maternel et paternel lors de la fécondation [10]. La première étape de la reprogrammation embryonnaire consiste à niveler le paysage chromatinien des deux pronoyaux parentaux (Figure 1B). Le génome du spermatozoïde subit une transformation drastique dans le cytoplasme de l’ovocyte. Il est progressivement décondensé par des histones acétylées apportées par l’ovocyte en remplacement des protamines, puis est soumis à une déméthylation globale de l’ADN impliquant des mécanismes actifs de déméthylation. Le génome maternel est quant à lui déméthylé de façon passive, principalement par dilution au cours des premiers clivages. Les épigénomes maternel et paternel sont aussi très différents en termes de marques d’histones. Par exemple, du fait du remplacement massif par les protamines, le génome paternel contient peu de marques d’histones telles que H3K9me2, H3K27me3 et H3K4me3, qui n’apparaissent qu’après la première réplication de l’ADN de l’embryon. Malgré ces différences d’organisation chromatinienne, les deux génomes parentaux sont fonctionnellement équivalents au moment de l’activation embryonnaire, à l’exception des gènes soumis à empreinte parentale, un sujet que nous développerons plus loin.
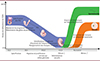 |
Figure 2 Changements majeurs, transcriptionnels et épigénétiques, au cours du développement des cellules germinales primordiales et de leur différentiation en gamètes matures. L’effacement de la méthylation de l’ADN dans les cellules germinales primordiales (PGC) se produit au cours de leur migration, puis est suivie d’une apposition de novo en fonction du sexe en pré et post natal dans les cellules mâles (vert) et femelles (orange) respectivement. La reprogrammation épigénétique des gamètes conduit également à l’apposition de grands domaines de la marque répressive H3K27me3 qui seront transmis à la descendance par l’ovocyte. |
La reprogrammation embryonnaire consiste ainsi en un effacement des profils liés à une identité gamétique qui culmine avec un état globalement hypométhylé du blastocyste à la fin du développement pré-implantatoire (environ 20 % de CpG méthylés) ; et ceci est vrai pour les deux lignages principaux, à savoir la masse cellulaire interne et le trophectoderme. La masse cellulaire interne diffère cependant avec la réactivation du chromosome X paternel inactif chez les rongeurs femelles [11]. C’est sur ce terrain épigénétiquement naïf que sont établis, après implantation et jusqu’à la gastrulation, les profils somatiques de méthylation de l’ADN, affectant jusqu’à 60 à 80 % de CpG méthylés, sous l’impulsion des DNMT de novo, DNMT3A et DNMT3B (Figure 1B) [8]. Cependant, le trophectoderme reste globalement hypométhylé en comparaison de l’épiblaste, et cette différence persiste entre le placenta et l’embryon jusqu’à la fin de la gestation. Cette asymétrie épigénétique des lignages embryonnaires et extra-embryonnaires concerne aussi l’enrichissement en marques d’histones, telles que H3K27me1, H3K27me2, H3K27me3 et H4K16ac [12,13].
Découverte de la reprogrammation épigénétique de cellules différenciées
Le voyage d’une cellule de la totipotence à la différenciation était considéré comme unidirectionnel jusqu’à la découverte, par les expériences de John Gurdon et Shinya Yamanaka, de la possibilité de reprogrammer des cellules somatiques.
Dès 1962, John Gurdon décrit le concept de reprogrammation cellulaire in vivo [14]. Il démontre que la spécialisation cellulaire n’est pas irréversible, en menant une expérience de transfert nucléaire chez l’amphibien Xenopus laevis. Des noyaux de cellules intestinales différenciées, transplantés dans des ovocytes énucléés, peuvent permettre le développement de têtards viables et fertiles, prouvant que le noyau des cellules différenciées contient toute l’information nécessaire pour générer un organisme entier. Cette découverte a montré que les marques épigénétiques liées à la spécialisation pouvaient être effacées ou réinitialisées, et que le cytoplasme de l’ovocyte jouait un rôle clé dans ce processus de reprogrammation [15]. John Gurdon mit 30 ans pour reproduire le transfert nucléaire chez les mammifères, car leur œuf est beaucoup plus petit que celui des amphibiens et le transfert des embryons nouvellement formés dans un utérus constituait alors un véritable exploit technique.
Quarante ans plus tard, Shinya Yamanaka fait un pas décisif en démontrant qu’il était possible, chez les mammifères, de reprogrammer in vitro des cellules somatiques différenciées en cellules souches pluripotentes induites (iPSC) [16]. Après avoir sélectionné 24 gènes candidats connus pour leur rôle dans le maintien de l’état de pluripotence cellulaire, Shinya Yamanaka teste tous ces facteurs un à un pour leur capacité à induire un état de pluripotence dans des cellules somatiques murines. Aucun facteur seul n’a cette capacité, mais en utilisant un cocktail de quatre facteurs de transcription (OCT3/4, SOX2, KLF4, c-MYC, appelés facteurs OKSM), Shinya Yamanaka réussit à convertir des fibroblastes adultes en cellules pluripotentes. Cette approche révolutionnaire montre que seules quelques instructions géniques suffisent pour qu’une cellule spécialisée retrouve la pluripotence et ainsi la capacité de se différencier en n’importe quel type cellulaire. Deux approches complémentaires ont permis de prouver la pluripotence complète des iPSC : la formation des trois feuillets germinaux après implantation sous la peau de souris receveuses et la contribution à générer un individu entier, y compris sa lignée germinale, après injection dans des blastocystes, [16].
Les découvertes de Gurdon et Yamanaka ont bouleversé notre compréhension de la différenciation cellulaire, en prouvant que la spécialisation d’une cellule n’était pas un processus irréversible, et que sous certaines conditions, une cellule mature pouvait être ramenée à un état pluripotent. Cette découverte a non seulement réécrit les manuels de biologie, mais a également ouvert des perspectives thérapeutiques immenses. Étant dérivées de cellules somatiques adultes, les cellules iPSC permettent de contourner les problèmes éthiques liés à l’utilisation des cellules souches embryonnaires et d’éviter les risques de rejet immunitaire. En utilisant les propres cellules d’un patient, il devient possible de produire des cellules souches personnalisées, ouvrant la voie à des traitements potentiels pour des maladies dégénératives, génétiques ou encore des lésions tissulaires. Cependant, malgré ces avancées, des défis demeurent, notamment le risque de tumeurs. En effet, l’utilisation du facteur oncogène c-MYC dans le processus de reprogrammation a suscité des inquiétudes quant à la stabilité génomique des iPSC. En 2021, une quarantaine d’essais cliniques étaient fondés sur l’utilisation d’iPSC, ce qui représente plus de 2 300 participants répartis dans le monde [17]. Le plus grand nombre d’essais cliniques est aujourd’hui ciblé sur le risque de malignité tumorale.
La lignée germinale chez les mammifères
Origine et développement
La fécondation d’un ovocyte par un spermatozoïde permet la création d’un organisme entier, viable et fertile, comprenant toutes les cellules différenciées nécessaires à son bon fonctionnement, ainsi que les cellules de sa lignée germinale. Ces cellules permettront à leur tour la création de la future génération et ainsi de suite. La lignée germinale peut en ce sens être considérée comme immortelle.
Chez l’embryon de souris, les PGC émergent d’une petite population de cellules (entre 10 et 15) au niveau de l’épiblaste proximal postérieur, en réponse à la signalisation BMP (bone morphogenic protein). Les PGC expriment le gène Prdm1 (aussi connu sous le nom de Blimp1 pour B lymphocyte-induced maturation protein 1) dès 6,25 jours de développement embryonnaire (E6,25), puis les gènes Prdm14 et Stella (aussi connu sous le nom de Dppa3 pour developmental pluripotency-associated 3) [18]. Les PGC vont ensuite migrer de la base de l’allantoïde1 (E8,5) jusqu’aux crêtes génitales, puis fortement proliférer et coloniser les futures gonades (E10,5 à E13,5) (Figure 2) [19]. La différenciation sexuelle des gonades en ovaires ou testicules a lieu à partir de E12, puis le devenir des PGC mâles et femelles diverge drastiquement. Chez les femelles, à E13,5, les PGC entrent en méiose I et resteront ensuite bloquées sous forme de follicules primaires en prophase 1 de méiose, jusqu’à reprendre leur progression à la puberté. Dans le cas des mâles, la méiose n’interviendra qu’après la naissance (Figure 2).
Reprogrammation des gamètes : changements transcriptionnels et chromatiniens
Lors de la spécification des cellules germinales primordiales, un programme transcriptionnel spécifique se met en place. D’un côté, les gènes de pluripotence sont réexprimés (Ssea1, Nanog, Oct4 et Sox2) et de l’autre le programme somatique est réprimé avec l’extinction transcriptionnelle de gènes tels que Hoxb1 (Homeobox) au profit de l’expression de Blimp1 (Figure 2) [19]. BLIMP1 est un répresseur transcriptionnel qui permet de réprimer les gènes de différenciation somatique lors de la reprogrammation de la lignée germinale.
Ces changements drastiques du transcriptome sont accompagnés d’une reprogrammation de l’épigénome tout aussi importante. Les cellules germinales primordiales sont très fortement déméthylées, progressivement entre E10,5 et E13,5 (Figure 2) [20,21]. Le génome atteint alors le plus bas niveau physiologique de méthylation, avec environ 6 % de CpG méthylés. Certaines régions spécifiques du génome se déméthylent plus lentement, tels que les gènes de la lignée germinale (germline genes), les séquences répétées et les régions de contrôle des gènes soumis à empreinte parentale [22,23]. La résistance des germline genes à la déméthylation est importante pour éviter une entrée précoce en méiose. La déméthylation se produit principalement par dilution passive lors des divisions cellulaires, mais TET1 pourrait aussi participer à la déméthylation active de quelques gènes clés de la différenciation germinale tardive et à la déméthylation globale du génome [22]. À contre-courant, certains éléments transposables, « jeunes » en terme d’évolution, échappent à la reprogrammation, et restent méthylés chez la souris et chez l’homme, vraisemblablement pour préserver l’intégrité du génome contre leur activité. Cette résistance à la reprogrammation peut être en partie expliquée par la présence de la marque répressive H3K9me3 à proximité de ces éléments [24,25].
La dynamique des marques des histones pendant cette période a été étudiée récemment, à l’échelle du génome, grâce à des techniques d’immuno-précipitation de la chromatine sur faible quantité. Les cellules germinales primordiales subissent une perte globale de H3K9me2 entre E8,5 et E9,5, et une forte accumulation de H3K27me3 à partir de E11,5. Un grand nombre de pics H3K27me3 et H3K9me3 sont différents dans les cellules germinales primordiales femelles et mâles, démontrant l’acquisition d’un paysage chromatinien spécifique au sexe [26]. De plus, dans les cellules germinales primordiales femelles, les gènes du chromosome X inactif sont réactivés progressivement à partir de E10,5 [27]. Ainsi, cette réinitialisation de l’épigénome des futurs gamètes pourrait avoir plusieurs finalités non exclusives : 1) l’activation des gènes de la lignée germinale ; 2) la mise en place de l’empreinte génomique parentale en fonction du sexe de l’individu ; et 3) l’effacement de mutations épigénétiques (ou épimutations) acquises, limitant leur transmission à la descendance (Figure 2).
L’empreinte génomique parentale
Définition et découverte
Les gènes soumis à empreinte génomique parentale se définissent par une expression mono-allélique en fonction de l’origine parentale, c’est-à-dire qu’un gène sera exprimé de façon mono-allélique soit uniquement à partir de l’allèle maternel, soit uniquement à partir de l’allèle paternel. Ce déterminisme allélique est contrôlé par la méthylation de l’ADN, différentiellement héritée des génomes parentaux. Les gènes soumis à empreinte parentale contiennent des motifs nucléotidiques de recrutement pour les protéines à doigt de zinc de la famille des KZFP (Kruppel-associated box (KRAB)associated zinc finger proteins), sensibles à la méthylation de leur site de liaison. Cette caractéristique des gènes soumis à empreinte parentale est nécessaire mais non suffisante pour les caractériser. Les motifs de recrutement pour les KZFP sont importants dans la transmission intergénérationnelle de l’empreinte, pour protéger la méthylation de l’ADN héritée des génomes parentaux (voir plus loin, le cycle de l’empreinte). Ainsi, bien que l’empreinte dépende d’une régulation épigénétique, via la méthylation de l’ADN héritée des gamètes, elle a finalement aussi un déterminisme génétique pour pouvoir être préservée dans l’embryon [28]. En effet, l’empreinte parentale canonique se maintient tout au long de la vie, quel que soit le sexe de l’individu. Ce processus concerne environ 140 gènes, soit environ 0,5 % des gènes annotés dans le génome.
L’empreinte génomique parentale célèbre les quarante ans de sa découverte cette année. En 1984, deux laboratoires indépendants, ceux de Azim Surani et Davor Solter, réalisent des expériences de transfert nucléaire dans des embryons murins pour étudier la contribution parentale au génome de l’embryon [29,30]. Dans un œuf fécondé énucléé de souris, les équipes transfèrent deux pronucleus, soit paternels, soit maternels, ou des deux origines, pour générer respectivement des embryons uniparentaux androgénotes2 et gynogénotes3, et des embryons biparentaux (comme contrôles) (Figure 3A). Contrairement aux embryons ayant reçu un pronoyau mâle et un pronoyau femelle, qui se développent normalement jusqu’à terme, les produits de conception (embryon et annexes extraembryonnaires) gynogénotes et androgénotes sont plus petits et dégénèrent en milieu de gestation [5]. Étonnamment donc, malgré leur diploïdie et leur contenu génétique identique (l’expérience étant réalisée sur une lignée de souris consanguine), ces embryons ne sont pas viables, et présentent de plus des phénotypes en miroir suivant l’origine parentale (Figure 3A) : l’embryon gynogénote a un développement relativement normal mais son trophoblaste est atrophié, tandis que l’embryon androgénote est très retardé avec un trophoblaste hyperplasique. Surani et Solter en conclueront que les deux génomes parentaux ne sont pas équivalents : ils portent une « empreinte » spécifique au sexe du parent donneur. Ainsi, la combinaison des génomes maternel et paternel via la reproduction sexuée est nécessaire pour le développement d’un embryon, en jouant des rôles complémentaires dans les tissus embryonnaires et extraembryonnaires.
 |
Figure 3 Découverte, cycle et mécanisme de l’empreinte parentale. A. Découverte de l’empreinte parentale par des expériences de transferts nucléaires. Illustration d’embryons et annexes extra-embryonnaires androgénotes, avec deux copies de génome paternel, ou gynogénotes, avec deux copies de génome maternel. (Illustrations des embryons avec la courtoisie du Prof M. Azim Surani). M : pronoyau maternel ; P : pronoyau paternel. B. Protection spécifique des locus soumis à l’empreinte parentale lors de la déméthylation globale du génome, juste après la fécondation. Représentation de la méthylation de l’ADN héritée du spermatozoïde en haut (vert) et de l’ovocyte en bas (orange). C. Exemple de la régulation d’un locus soumis à l’empreinte parentale, Igf2-H19, localisé sur le chromosome 7 de la souris. Le gène Igf2 est exprimé uniquement à partir de l’allèle paternel et le gène H19 uniquement à partir de l’allèle maternel. La région de contrôle de l’empreinte (ICR) correspond à une région différentiellement méthylée entre les deux allèles parentaux, transmise méthylée uniquement dans le génome paternel. En absence de méthylation sur l’ICR, la protéine insulatrice CTCF se fixe sur l’allèle maternel et permet aux boucles de chromatine formées d’isoler le gène Igf2 de ses enhancers. Sur l’allèle paternel, ces enhancers peuvent contacter le gène Igf2, en absence de CTCF. |
Par la suite, Bruce Cattanach étudia un grand nombre de translocations réciproques chez la souris, générant des régions chromosomiques uniquement maternelles ou paternelles [31]. Ce panel de disomies uniparentales permit à Bruce Cattanach de cartographier des régions dont les effets phénotypiques, notamment sur la viabilité et la croissance, dépendent de l’origine parentale. En 1991, les premiers gènes soumis à l’empreinte parentale sont décrits : Igf2R, H19 et Igf2. Le phénotype de leur invalidation peut donc être étudié en fonction de l’origine parentale de la délétion dans des modèles murins [5]. Les années suivantes ont été marquées par la découverte de nombreux locus soumis à empreinte parentale, étudiés grâce à des modèles d’invalidation génique chez la souris. De nombreux gènes soumis à l’empreinte ont également été découverts chez l’homme, à travers l’étude des maladies à effet parental associées [32,33]. Enfin, il est rapidement apparu que ce phénomène de non-équivalence fonctionnelle parentale était lié à des profils asymétriques de méthylation transmis par le spermatozoïde et par l’ovocyte.
Le cycle de l’empreinte
Le cycle de l’empreinte parentale recouvre les phases successives d’effacement et d’établissement à chaque génération pour que les gamètes transmettent uniquement les marques en adéquation avec le sexe de l’individu et non celles héritées des parents. Ainsi, l’empreinte parentale dans sa forme canonique est apposée via la méthylation de l’ADN lors de la gamétogenèse mâle et femelle sous l’action de DNMT3A, stimulée par DNMT3L (Figure 2) [5,34]. Les profils de méthylation des gamètes parentaux sont globalement différents, ainsi que leur dynamique d’établissement lors de la gamétogenèse dans les deux sexes. La méthylation du génome du futur spermatozoïde est acquise avant la naissance, dans les prospermatogonies qui sont issues des PGC, et affecte globalement tout le génome, atteignant 80 % de CpG méthylés. Le génome maternel est quant à lui méthylé après la naissance dans les ovocytes en phase de croissance, principalement dans le corps des gènes qui sont exprimés dans l’ovocyte, cumulant environ 55 % de CpG méthylés. Ces profils contrastés entre spermatozoïde et ovocyte générent de multiples régions de méthylation différentielle (DMR pour differentially methylated region), qui sont transmises par les gamètes à la génération suivante.
Cependant, l’embryon subit une reprogrammation globale de son épigénome, incluant une déméthylation de l’ADN après la fécondation (Figure 3B). En conséquence, alors que de multiples DMR sont héritées, la reprogrammation épigénétique permet d’effacer ces différences parentales dans l’embryon, à l’exception des régions soumises à l’empreinte, ciblées par des mécanismes de protection qui leur permettent de conserver la mémoire des profils alléliques de méthylation parentale (Figure 3B). Le seul mécanisme connu permettant de protéger l’empreinte de la reprogrammation repose sur le recrutement de facteurs de liaison à la chromatine qui vont protéger la méthylation des CpG au niveau des locus soumis à l’empreinte lors de la déméthylation globale du génome. Les protéines à doigt de zinc ZFP57 et ZFP445 reconnaissent leur séquence nucléotidique de référence uniquement lorsqu’elle est méthylée [5]. Ces KZFP permettent le recrutement du répresseur KAP1 (KRAB-associated corepressor) qui favorise la triméthylation de H3K9me3 (via SETDB1), et de DNMT1 et UHRF1 sur les régions régulatrices méthylées des locus soumis à empreinte [35]. La présence de DNMT1 et UHRF1 est requise pour le maintien de la méthylation, alors que le reste des génomes parentaux est déméthylé. Pour conserver l’asymétrie de méthylation entre les allèles parentaux, l’allèle hérité non méthylé doit aussi être protégé de la reméthylation du génome qui a lieu dans l’épiblaste après implantation. La présence des marques bivalentes H3K4me3 et H3K27me3 au niveau de l’allèle non méthylé pourrait jouer ce rôle de protection mais le mécanisme reste encore peu caractérisé (Figure 1B et Figure 3B) [36]. Les marques de méthylation liées à l’empreinte sont ensuite lues dans le placenta et les lignages somatiques. Dans les PGC, l’empreinte est effacée afin de réapposer une nouvelle empreinte, spécifique au sexe de l’individu pour sa propre descendance. Il existe donc un cycle crucial d’effacement et de réapposition dans la lignée germinale, de protection dans l’embryon préimplantatoire, puis de lecture des marques permettant l’expression mono-allélique des gènes soumis à empreinte, en fonction de leur origine parentale.
Mécanismes de régulation des gènes soumis à l’empreinte parentale
Les gènes soumis à empreinte sont communément regroupés en grands domaines chromosomiques, régulés par une ou plusieurs DMR. Ces DMR peuvent être de deux types : les DMR gamétiques, méthylées lors de la gamétogenèse, et qui peuvent se comporter comme des centres de contrôle de l’empreinte (ICR pour imprinting control region), et des DMR somatiques, acquises au cours du développement de l’individu. Les ICR peuvent contrôler l’expression mono-allélique de tous les gènes soumis à l’empreinte d’un locus, à plusieurs kilobases de distance. Toutes les ICR connues à ce jour sont des DMR gamétiques, avec un rôle prédominant du génome maternel : chez la souris, la méthylation de 18 ICR sur les 21 répertoriées est acquise dans l’ovocyte contre seulement 3 dans le spermatozoïde. Ainsi, la régulation des locus soumis à l’empreinte parentale concerne des gènes en clusters, contrôlés par une ICR porteuse de la méthylation différentielle parentale. Elle implique de plus un contrôle par les marques d’histones et pardes ARN non codants, ainsi que par des protéines insulatrices de la chromatine, telles que CTCF (CCCTC-binding factor), responsables de la formation de boucles de chromatine entre enhancers et promoteurs. Un des mécanismes les mieux disséqués concerne le locus H19-Igf2, soumis à une empreinte paternelle (Figure 3C) [37]. Le gène H19 produit un ARN non codant exprimé pendant le développement embryonaire, exclusivement à partir de l’allèle maternel. Son voisin, le gène Igf2 (insulin-like growth factor 2), code un facteur de croissance exprimé uniquement à partir de l’allèle paternel [38–40]. L’ICR du locus H19-Igf2 se situe à environ 2 à 4 kb en amont du gène H19, possède plusieurs sites de fixation pour la protéine insulatrice CTCF, et porte une méthylation héritée du spermatozoïde. CTCF, qui est sensible à la méthylation de l’ADN, ne peut donc se fixer que sur l’allèle maternel déméthylé, isolant ainsi les séquences enhancers des promoteurs d’Igf2, et les rendant disponibles pour le promoteur d’H19. De plus, l’ARN non codant H19 joue le rôle de plateforme de recrutement de répresseurs transcriptionnels de Igf2 en cis. Ceci permet d’aboutir à une expression de H19 exclusive sur l’allèle maternel. Sur l’allèle paternel, en absence de CTCF, les enhancers peuvent contacter les séquences promotrices d’Igf2, permettant son expression tandis que le promoteur d’H19 se retrouve méthylé. Ces différents acteurs permettent la régulation fine et souvent complexe de nombreux locus soumis à empreinte, bien que chaque cluster semble dépendre de mécanismes propres.
Syndromes humains liés à l’empreinte parentale
Au cours du développement, les gènes régulés par l’empreinte parentale sont nécessaires pour la croissance de l’embryon et du nouveau-né, et le développement du placenta. Ces gènes sont également exprimés au cours de la vie adulte et ont des fonctions très importantes pour le cerveau, comme la neurogenèse, le sommeil, le rythme circadien, la mémoire, ainsi que certains comportements sociaux et de soins maternels, tels que l’allaitement. Chez l’homme, des défauts génétiques de certains locus soumis à l’empreinte se traduisent par des syndromes lourds et opposés selon l’origine parentale de l’allèle touché. Les principales causes de maladie sont dues à des disomies uniparentales, et des pertes d’empreinte dues à des mutations génétiques ou à des épimutations (Tableau I).
Le locus H19-IGF2 localisé sur le chromosome 11 humain est associé aux syndromes développementaux de Beckwith-Wiedemann et de Silver-Russell [41]. Le syndrome de Beckwith-Wiedemann entraîne une surcroissance prénatale et/ou postnatale, une hypertrophie de la langue, des défauts de la paroi abdominale, une surcroissance du placenta et une prédisposition à des tumeurs embryonnaires. Il existe de nombreux défauts moléculaires, comme des disomies parentales paternelles, c’est-à-dire la présence des deux copies du chromosome 11 paternel, conduisant à une absence d’expression de H19 et une double dose d’IGF2. Ces disomies sont détectées chez 20 % des patients. Il existe également des cas de gain de méthylation de l’allèle maternel dans 5 à 10 % des cas (épimutation). Cette situation conduit également à une surexpression d’IGF2 pendant le développement embryonnaire, entraînant une croissance excessive généralisée.
Contrairement au syndrome de Beckwith-Wiedemann, le syndrome de Silver-Russell est associé à un retard de croissance. Ce syndrome est causé par un taux d’expression d’IGF2 trop bas. Pour plus de 50 % des patients souffrant de ce syndrome, une perte de la méthylation est constatée sur l’allèle paternel de l’ICR d’H19, ce qui bloque l’expression d’IGF2 et conduit à une expression biallelique de H19 [42]. Il existe de nombreuses autres maladies associées à des gènes soumis à l’empreinte parentale (Tableau I), illustrant l’importance de ce mécanisme de régulation épigénétique pour le développement embryonnaire mais également postnatal.
Les syndromes humains liés à des défauts génétiques et/ou épigénétiques dans des locus soumis à l’empreinte parentale. Ce Tableau décrit les principaux syndromes humains liés à des gènes soumis à l’empreinte parentale avec leur localisation chromosomique, leurs principales caractéristiques et leurs origines épigénétiques et génétiques.
De nouvelles formes d’empreinte parentale non canonique
Les gènes soumis à empreinte parentale canonique se caractérisent par une méthylation ubiquitaire portée par une ICR, et une expression mono-allélique des gènes associés dans tous les tissus (où ils sont exprimés) et tout au long de la vie. Cependant, les gènes soumis à empreinte les plus récemment mis en évidence montrent souvent une forme non canonique d’empreinte, avec des ICR moins robustes, qui ne conservent la méthylation différentielle héritée des gamètes parentaux que sur une courte période développementale ou dans un tissu spécifique. Ces ICR sont sujettes à regagner de la méthylation sur l’allèle initialement hérité avec un état non méthylé. Cependant, pour les formes transitoires qui n’existent qu’au début du développement embryonnaire, l’effet de l’empreinte parentale peut perdurer, comme démontré pour le locus Zdbf2 (zinc finger DBF-type containing 2) [43]. L’ICR de ce locus est méthylée maternellement, conduisant à l’expression mono-allélique paternel d’un transcript alternatif de Zdbf2, uniquement pendant le développement préimplantatoire. Bien que l’ICR disparaisse à l’implantation par reméthylation de l’allèle paternel, cette brève empreinte est nécessaire et suffisante pour programmer l’expression mono-allélique de Zdbf2 après la naissance dans l’axe hypothalamo-hypophysaire, régulant la prise alimentaire et la croissance du nouveau-né chez la souris [44].
Récemment, en 2017, une nouvelle forme d’empreinte parentale non canonique a été découverte, dans laquelle la marque gamétique n’est pas dépendante de la méthylation de l’ADN, mais de la marque H3K27me3, héritée systématiquement de l’ovocyte. En effet, le paysage chromatinien de l’ovocyte se distingue par de larges domaines de H3K27me3, qui sont transmis au zygote et persistent durant le développement préimplantatoire, conduisant à une asymétrie avec le génome paternel. Les 76 gènes répertoriés dans ces domaines de H3K27me3 maternels sont exprimés transitoirement de façon mono-allélique paternelle [6]. Cependant, l’enrichissement maternel asymétrique en H3K27me3 subit une dilution dans les cellules de la masse interne. Il n’est plus détectable dans l’épiblaste de l’embryon post-implantoire à E6,5, mais perdure pour quelques rares gènes dans le placenta et semble important pour le développement de ce tissu [45,46].
Conclusion
La reprogrammation épigénétique désigne le processus par lequel les marques de la chromatine, telles que la méthylation de l’ADN et les modifications des histones, sont effacées et/ou redistribuées, permettant de faire « table rase » de l’épigénome d’une cellule. Durant le développement, la reprogrammation épigénétique accompagne l’organisation du génome de l’embryon. Elle est également cruciale pour la formation de gamètes fonctionnels, débarrassés des éventuelles épimutations acquises au cours du développement et dotés de marques chromatiniennes spécifiques au sexe, nécessaires au développement de la génération suivante. En effet, la réinitialisation de l’empreinte parentale est indispensable au développement du futur embryon qui est compromis en l’absence de ce contrôle épigénétique parental, dans ses formes canonique ou non canoniques. Récemment, en plus de la méthylation de l’ADN, la transmission de la marque H3K27me3 par l’ovocyte a été montrée comme essentielle pour la génération suivante. D’autres marques épigénétiques sont probablement impliquées dans la transmission intergénérationnelle d’informations, et leur identification constitue un terrain important de la recherche dans ce domaine.
Comprendre les mécanismes impliqués dans la reprogrammation est vital, car ils influencent non seulement le développement normal mais également la santé des descendants. La capacité à reprogrammer un noyau somatique a également révolutionné notre compréhension du développement, de la biologie et de l’avenir de la médecine. En effet, les premiers clonages animaux ont été réalisés grâce au principe de reprogrammation d’un noyau d’une cellule différenciée. La possibilité de dédifférencier des cellules de patients en iPSC permet de contourner les risques de rejet et ouvre un très grand nombre d’applications médicales. Les recherches sur la lignée germinale sont également cruciales pour mieux comprendre les interactions entre reprogrammation épigénétique, fertilité et syndromes développementaux. La reprogrammation épigénétique est un domaine fascinant de la biologie, fondamentale et appliquée, qui n’a pas encore révélé tous ses rouages.
Remerciements
Le travail au laboratoire est soutenu par une subvention ATIP-Avenir du CNRS et de l’Inserm, par l’amorçage Jeune équipe de la Fondation pour la recherche médicale (FRM - AJE202005011598) et par l’Agence nationale de la recherche dans le cadre du programme « Investissements d’avenir » sous la référence ANR-16-IDEX-0006. C. Roidor bénéficie d’une bourse de doctorat de la Ligue nationale contre le cancer.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Allantoïde : organe fœtal dont la partie extra-embryonnaire dégénère durant la gestation. La partie intra-embryonnaire donne la future vessie.
Embryon androgénote : embryon se développant à partir d’un œuf contenant deux pronucleus paternels.
Embryon gynogénote: embryon se développant à partir d’un œuf contenant deux pronucleus maternels.
Références
- Waddington CH. The Epigenotype. Int J Epidemiol 2012 ; 41 : 10–3. [CrossRef] [PubMed] [Google Scholar]
- Goldberg AD, Allis CD, Bernstein E. Epigenetics: A Landscape Takes Shape. Cell 2007 ; 128 : 635–8. [CrossRef] [PubMed] [Google Scholar]
- Johnson MH, Cohen J. Reprogramming rewarded: the 2012 Nobel prize for Physiology or Medicine awarded to John Gurdon and Shinya Yamanaka. Reprod Biomed Online 2012 ; 25 : 549–50. [CrossRef] [PubMed] [Google Scholar]
- Hajkova P, Ancelin K, Waldmann T, et al. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 2008 ; 452 : 877–81. [CrossRef] [PubMed] [Google Scholar]
- Tucci V, Isles AR, Kelsey G, et al. Genomic Imprinting and Physiological Processes in Mammals. Cell 2019 ; 176 : 952–65. [CrossRef] [PubMed] [Google Scholar]
- Inoue A, Jiang L, Lu F, et al. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 2017 ; 547 : 419–24. [CrossRef] [PubMed] [Google Scholar]
- Ea V, Baudement M-O, Lesne A, et al. Contribution of Topological Domains and Loop Formation to 3D Chromatin Organization. Genes 2015 ; 6 : 734–50. [CrossRef] [PubMed] [Google Scholar]
- Greenberg MVC, Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019 ; 20 : 590–607. [CrossRef] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S. The Yin and Yang of Histone Marks in Transcription. Annu Rev Genomics Hum Genet 2021 ; 22 : 147–70. [CrossRef] [PubMed] [Google Scholar]
- Surani MA, Hayashi K, Hajkova P. Genetic and Epigenetic Regulators of Pluripotency. Cell 2007 ; 128 : 747–62. [CrossRef] [PubMed] [Google Scholar]
- Borensztein M, Okamoto I, Syx L, et al. Contribution of epigenetic landscapes and transcription factors to X-chromosome reactivation in the inner cell mass. Nat Commun 2017 ; 8 : 1–14. [CrossRef] [PubMed] [Google Scholar]
- Dahl JA, Reiner AH, Klungland A, et al. Histone H3 Lysine 27 Methylation Asymmetry on Developmentally-Regulated Promoters Distinguish the First Two Lineages in Mouse Preimplantation Embryos. PLoS ONE 2010 ; 5 : e9150. [CrossRef] [PubMed] [Google Scholar]
- Gupta A, Guerin-Peyrou TG, Sharma GG, et al. The Mammalian Ortholog of Drosophila MOF That Acetylates Histone H4 Lysine 16 Is Essential for Embryogenesis and Oncogenesis. Mol Cell Biol 2008 ; 28 : 397–409. [CrossRef] [PubMed] [Google Scholar]
- Gurdon JB. The Developmental Capacity of Nuclei taken from Intestinal Epithelium Cells of Feeding Tadpoles. Development 1962 ; 10 : 622–40. [CrossRef] [Google Scholar]
- Gurdon JB, Melton DA. Nuclear Reprogramming in Cells. Science 2008 ; 322 : 1811–5. [CrossRef] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006 ; 126 : 663–76. [CrossRef] [PubMed] [Google Scholar]
- Ilic D, Ogilvie C. Pluripotent Stem Cells in Clinical Setting—New Developments and Overview of Current Status. Stem Cells 2022 ; 40 : 791–801. [CrossRef] [PubMed] [Google Scholar]
- Saitou M, Hayashi K. Mammalian in vitro gametogenesis. Science 2021 ; 374 : eaaz6830. [CrossRef] [PubMed] [Google Scholar]
- Kurimoto K, Saitou M. Germ cell reprogramming. 1st ed. Amsterdam : Elsevier Inc., 2019. pp. 91–125. [Google Scholar]
- Kobayashi H, Sakurai T, Miura F, et al. High-resolution DNA methylome analysis of primordial germ cells identifies gender-specific reprogramming in mice. Genome Res 2013 ; 23 : 616–27. [CrossRef] [PubMed] [Google Scholar]
- Seisenberger S, Andrews S, Krueger F, et al. The Dynamics of Genome-wide DNA Methylation Reprogramming in Mouse Primordial Germ Cells. Mol Cell 2012 ; 48 : 849–62. [CrossRef] [PubMed] [Google Scholar]
- Hill PWS, Leitch HG #1, Requena CE#1, et al. Epigenetic reprogramming enables the primordial germ cell-to-gonocyte transition Europe PMC Funders Group. Nature 2018 ; 555 : 392–6. [CrossRef] [PubMed] [Google Scholar]
- Guibert S, Forne T, Weber M. Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res 2012 ; 22 : 633–41. [CrossRef] [PubMed] [Google Scholar]
- Karimi MM, Goyal P, Maksakova IA, et al. DNA Methylation and SETDB1/H3K9me3 Regulate Predominantly Distinct Sets of Genes, Retroelements, and Chimeric Transcripts in mESCs. Cell Stem Cell 2011 ; 8 : 676–87. [CrossRef] [PubMed] [Google Scholar]
- Liu S, Brind’Amour J, Karimi MM, et al. Setdb1 is required for germline development and silencing of H3K9me3-marked endogenous retroviruses in primordial germ cells. Genes Dev 2014 ; 28 : 2041–55. [CrossRef] [PubMed] [Google Scholar]
- Huang T-C, Wang Y-F, Vazquez-Ferrer E, et al. Sex-specific chromatin remodelling safeguards transcription in germ cells. Nature 2021 ; 600 : 737–42. [CrossRef] [PubMed] [Google Scholar]
- BiorXiv doi.org/10.1101/2023.04.25.532252 [Google Scholar]
- Juan AM, Bartolomei MS. Evolving imprinting control regions: KRAB zinc fingers hold the key. Genes Dev 2019 ; 33 : 1–3. [CrossRef] [PubMed] [Google Scholar]
- Surani MAH, Barton SC, Norris ML. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 1984 ; 308 : 548–50. [CrossRef] [PubMed] [Google Scholar]
- McGrath J, Solter D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 1984 ; 37 : 179–83. [CrossRef] [PubMed] [Google Scholar]
- Cattanach BM, Kirk M. Differential activity of maternally and paternally derived chromosome regions in mice. Nature 1985 ; 315 : 496–8. [CrossRef] [PubMed] [Google Scholar]
- Nicholls RD, Knoll JHM, Butler MG, et al. Genetic imprinting suggested by maternal heterodisomy in non-deletion Prader-Willi syndrome. Nature 1989 ; 342 : 281–5. [CrossRef] [PubMed] [Google Scholar]
- Williams CA, Zori RT, Stone JW, et al. Maternal origin of 15q11-13 deletions in Angelman syndrome suggests a role for genomic imprinting. Am J Med Genet 1990 ; 35 : 350–3. [CrossRef] [PubMed] [Google Scholar]
- Bourc’his D, Xu GL, Lin CS, et al. Dnmt3L and the establishment of maternal genomic imprints. Science 2001 ; 294 : 2536–9. [CrossRef] [PubMed] [Google Scholar]
- Messerschmidt DM. Should I stay or should I go: protection and maintenance of DNA methylation at imprinted genes. Epigenetics 2012 ; 7 : 969–75. [CrossRef] [PubMed] [Google Scholar]
- Maupetit-Méhouas S, Montibus B, Nury D, et al. Imprinting control regions (ICRs) are marked by mono-allelic bivalent chromatin when transcriptionally inactive. Nucleic Acids Res 2015 ; 44 : 621. [Google Scholar]
- Gabory A, Jammes H, Dandolo L. The H19 locus: Role of an imprinted non-coding RNA in growth and development. BioEssays 2010 ; 32 : 473–80. [CrossRef] [PubMed] [Google Scholar]
- Ferguson-Smith AC, Sasaki H, Cattanach BM, et al. Parental-origin-specific epigenetic modification of the mouse H19 gene. Nature 1993 ; 362 : 751–5. [CrossRef] [PubMed] [Google Scholar]
- Bartolomei MS, Webber AL, Brunkow ME, et al. Epigenetic mechanisms underlying the imprinting of the mouse H19 gene. Genes Dev 1993 ; 7 : 1663–73. [CrossRef] [PubMed] [Google Scholar]
- DeChiara TM, Efstratiadis A, Robertson EJ. A growth-deficiency phenotype in heterozygous mice carryng an insulin-like growth factor II gene disrupted by targeting. Nature 1990 ; 374 : 685–9. [Google Scholar]
- Brioude F, Kalish JM, Mussa A, et al. Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol 2018 ; 14 : 229–49. [CrossRef] [PubMed] [Google Scholar]
- Russo S, Calzari L, Mussa A, et al. A multi-method approach to the molecular diagnosis of overt and borderline 11p15.5 defects underlying Silver-Russell and Beckwith-Wiedemann syndromes. Clin Epigenetics 2016 ; 8 : 23. [CrossRef] [PubMed] [Google Scholar]
- Greenberg MVC, Glaser J, Borsos M, et al. Transient transcription in the early embryo sets an epigenetic state that programs postnatal growth. Nat Genet 2017 ; 49 : 110–8. [CrossRef] [PubMed] [Google Scholar]
- Glaser J, Iranzo J, Borensztein M, et al. The imprinted Zdbf2 gene finely tunes control of feeding and growth in neonates. eLife 2022 ; 11 : e65641. [CrossRef] [PubMed] [Google Scholar]
- Inoue A, Jiang L, Lu F, et al. Genomic imprinting of Xist by maternal H3K27me3. Genes Dev 2017 ; 31 : 1927–32. [CrossRef] [PubMed] [Google Scholar]
- Matoba S, Kozuka C, Miura K, et al. Noncanonical imprinting sustains embryonic development and restrains placental overgrowth. Genes Dev 2022 ; genesdev ;gad.349390.122v1. [PubMed] [Google Scholar]
Liste des tableaux
Les syndromes humains liés à des défauts génétiques et/ou épigénétiques dans des locus soumis à l’empreinte parentale. Ce Tableau décrit les principaux syndromes humains liés à des gènes soumis à l’empreinte parentale avec leur localisation chromosomique, leurs principales caractéristiques et leurs origines épigénétiques et génétiques.
Liste des figures
|
Figure 1 Le cycle de programmation et reprogrammation épigénétique au cours du développement murin. A. Les grandes étapes d’apposition et effacement des marques épigénétiques chez la souris. B. Représentation des niveaux globaux de méthylation de l’ADN des gamètes à la souris adulte. TE : trophectoderme ; MCI : masse cellulaire interne ; PE : endoderme primitif ; ExE : ectoderme extra-embryonnaire ; VE : endoderme viscéral ; Epi : épiblaste ; E0,5 : 0,5 jours de développement embryonnaire. |
| Dans le texte | |
|
Figure 2 Changements majeurs, transcriptionnels et épigénétiques, au cours du développement des cellules germinales primordiales et de leur différentiation en gamètes matures. L’effacement de la méthylation de l’ADN dans les cellules germinales primordiales (PGC) se produit au cours de leur migration, puis est suivie d’une apposition de novo en fonction du sexe en pré et post natal dans les cellules mâles (vert) et femelles (orange) respectivement. La reprogrammation épigénétique des gamètes conduit également à l’apposition de grands domaines de la marque répressive H3K27me3 qui seront transmis à la descendance par l’ovocyte. |
| Dans le texte | |
|
Figure 3 Découverte, cycle et mécanisme de l’empreinte parentale. A. Découverte de l’empreinte parentale par des expériences de transferts nucléaires. Illustration d’embryons et annexes extra-embryonnaires androgénotes, avec deux copies de génome paternel, ou gynogénotes, avec deux copies de génome maternel. (Illustrations des embryons avec la courtoisie du Prof M. Azim Surani). M : pronoyau maternel ; P : pronoyau paternel. B. Protection spécifique des locus soumis à l’empreinte parentale lors de la déméthylation globale du génome, juste après la fécondation. Représentation de la méthylation de l’ADN héritée du spermatozoïde en haut (vert) et de l’ovocyte en bas (orange). C. Exemple de la régulation d’un locus soumis à l’empreinte parentale, Igf2-H19, localisé sur le chromosome 7 de la souris. Le gène Igf2 est exprimé uniquement à partir de l’allèle paternel et le gène H19 uniquement à partir de l’allèle maternel. La région de contrôle de l’empreinte (ICR) correspond à une région différentiellement méthylée entre les deux allèles parentaux, transmise méthylée uniquement dans le génome paternel. En absence de méthylation sur l’ICR, la protéine insulatrice CTCF se fixe sur l’allèle maternel et permet aux boucles de chromatine formées d’isoler le gène Igf2 de ses enhancers. Sur l’allèle paternel, ces enhancers peuvent contacter le gène Igf2, en absence de CTCF. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.