")
")
| Issue |
Med Sci (Paris)
Volume 41, Number 6-7, Juin-Juillet 2025
|
|
|---|---|---|
| Page(s) | 570 - 577 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2025095 | |
| Published online | 7 juillet 2025 | |
Le méthylome ou l’avenir de la neuro-oncologie ?
DNA methylation profiling or the future of neuro-oncology?
1
Département de pathologie, CHU d’Angers, France
2
Centre de recherche en cancérologie et immunologie intégrée de Nantes-Angers (CRCI2NA), Inserm U1307, CNRS UMR6075, Université de Nantes/Angers, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Le diagnostic des tumeurs du système nerveux central repose à la fois sur les aspects histopathologiques et les anomalies moléculaires des tumeurs. La caractérisation moléculaire s’est enrichie de l’analyse des profils de méthylation. Le méthylome, reflet de la cellule d’origine de la tumeur et des modifications épigénétiques subies pendant la tumorigenèse, permet, grâce à un algorithme d’intelligence artificielle, de classer les tumeurs du système nerveux central. Sa mise en œuvre a révolutionné la neuro-oncologie, tant sur le plan diagnostique que pronostique, notamment en pédiatrie. Pour l’instant, l’analyse du méthylome nécessite de partager les données avec le créateur de l’algorithme. Cette analyse étant appelée à se généraliser en oncologie, des solutions alternatives telles que le séquençage par nanopore, sont à développer.
Abstract
Diagnosis of central nervous system tumors is based on the histopathologic features and molecular alterations. For certain entities, this histomolecular diagnosis now includes DNA methylation profiles. Methylation profiles reflect the cell of origin and epigenetic modifications that occur during tumorigenesis and may help classify central nervous system tumors via artificial intelligence algorithms. This powerful approach to tumor classification has revolutionized the field of neuro-oncology, allowing more reliable diagnosis and prognostic assessment, particularly for pediatric neoplasms. The analysis of methylation profiles currently requires data to be shared with the developer of the AI algorithms. As DNA methylation profiling may be useful in a growing number of cancers, local alternatives, such as those based on the nanopore technology, need to be developed.
© 2025 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.

Vignette (© Alix Fontaine).
Les tumeurs du système nerveux central sont à l’origine d’une importante morbi-mortalité [1]. Les gliomes sont les tumeurs primitives du système nerveux central les plus fréquentes, avec notamment le glioblastome (3 481 nouveaux cas par an en France), qui est la forme la plus agressive des tumeurs gliales [2]. Chez l’enfant, les gliomes malins prédominent aux côtés des tumeurs embryonnaires, tout particulièrement des médulloblastomes (tumeurs agressives du cervelet) [1]. Les tumeurs du système nerveux central de l’enfant sont beaucoup plus variées que celles de l’adulte et leur diagnostic est souvent plus difficile. Les traitements en neuro-oncologie ont peu évolué au cours des 20 dernières années et la survie globale des patients atteints d’un glioblastome reste de 15 mois après traitement [3].
L’Organisation mondiale de la santé (OMS) classe les tumeurs du système nerveux central selon : 1) l’âge de survenue (pédiatrique versus adulte), 2) l’origine cellulaire supposée de la tumeur (gliale, neuronale, etc.), 3) l’aspect des cellules au microscope (histopathologie), 4) l’expression de marqueurs de différenciation (détectée par immunohistochimie), 5) l’agressivité de la tumeur (selon un classement en grades allant de 1 à 4), et 6) les altérations moléculaires clés (anomalies des chromosomes, mutations, transcrits de fusion) [4].
La classification de l’OMS des tumeurs du système nerveux central est un référentiel qui évolue au fil du temps. Jusqu’en 2016, cette classification était basée sur les caractéristiques morphologiques et immunohistochimiques des tumeurs [5]. Les progrès en biologie moléculaire, tout particulièrement l’avènement du séquençage à haut débit (next generation sequencing), ont permis une caractérisation plus fine de nombreuses tumeurs. La classification de l’OMS de 2016 alliait, pour la première fois, les aspects histopathologiques et les caractéristiques moléculaires, introduisant la notion de « classification histomoléculaire » et de « diagnostic intégré ». Ce diagnostic en couches, ou en strates, prend en compte les données clinico-radiologiques, histopathologiques et moléculaires [6]. Il est plus objectif que la seule appréciation des cellules tumorales au microscope et représente une avancée majeure en neuro-oncologie.
Les progrès de la biologie moléculaire ont permis d’explorer une autre caractéristique des tumeurs : leur profil épigénétique. Le diagnostic intégré comprend maintenant un niveau supplémentaire (après les données cliniques, le type histologique, le grade et l’altération génétique) : la classe de méthylation. La dernière classification de l’OMS, éditée en 2021, intègre dans la démarche diagnostique, pour certains types tumoraux, les données du méthylome [4]. Cette analyse épigénétique fait partie des critères désirables, voire essentiels, pour porter un diagnostic en neuro-oncologie. La classification de l’OMS met ainsi en exergue la caractérisation moléculaire des tumeurs, parfois au détriment des aspects histopathologiques.
L’objectif de cette synthèse est d’exposer les principes du méthylome, sa place dans le diagnostic des tumeurs du système nerveux central, ses avantages mais aussi ses limites et, enfin, l’avenir des analyses épigénétiques en neuro-oncologie.
La méthylation de l’ADN
Un mécanisme de régulation dynamique et transmissible
La méthylation de l’ADN est un puissant mécanisme de régulation de l’expression des gènes. Il s’agit d’un processus biologique au cours duquel des groupements méthyle (CH3) sont ajoutés à ou retirés de la molécule d’ADN par les enzymes méthyltransférases (DNMT pour DNA methyltransferases) et des méthylcytosine dioxygénases telles que TET2 [7]. Dans les deux cas, l’ajout des groupements méthyle a lieu sur les bases cytosines reliées par une liaison phosphate à une base guanine (CpG) du même brin d’ADN [7] . La méthylation peut modifier l’activité d’un segment d’ADN sans en modifier la séquence nucléotidique. La méthylation des îlots CpG, qui sont des zones à forte concentration de dinucléotides CpG, au sein du promoteur d’un gène entraîne généralement la répression de sa transcription [8]. Par contre, la méthylation des îlots CpG localisés dans le corps du gène (méthylation intragénique), c’est-à-dire dans les introns ou exons, entraîne soit une activation, soit une répression de l’expression du gène [9].
Chez les mammifères, la méthylation de l’ADN est essentielle au développement normal et est associée à des processus biologiques clés, tels que l’embryogenèse ou le vieillissement [7–10] (→).
(→) Voir m/s n° 8-9, 2008, page 731
Il s’agit d’un phénomène dynamique et transmissible qui répond à des stimuli et permet une adaptation rapide de l’organisme vivant à son environnement [11]. Des modifications du méthylome sont observées en cas de stress nutritionnel, psychique ou d’exposition à des toxiques [12, 13]. Ces modifications épigénétiques aboutissent à des changements de comportement, à l’acquisition de nouveaux phénotypes, et favorisent le développement de certaines maladies [12, 13].
Une carte d’identité cellulaire
Le méthylome complet de l’ADN humain est disponible depuis 2023 [14]. C’est un outil utilisé aussi bien pour détecter des expositions environnementales que pour comprendre la physiopathologie d’une maladie ou identifier l’origine de cellules tumorales [15, 16]. La méthylation permet la spéciation cellulaire [7]. Chaque cellule acquiert, au cours de l’embryogenèse, un profil de méthylation stable qui est propre à son organe d’origine. Ce profil peut être caractéristique d’une région de l’organe en question ou d’un sous-type cellulaire [17]. Ceci est particulièrement vrai dans le système nerveux central, où les profils de méthylation permettent de distinguer les différents types cellulaires (gliaux, neuronaux, épendymaires, etc.) et leur localisation anatomique (hémisphères cérébraux, tronc cérébral/cervelet, moelle épinière) [17, 18]. Les modifications épigénétiques ultérieures acquises au cours de la tumorigenèse altèrent relativement peu cette signature [16]. La méthylation de l’ADN est principalement déterminée par l’origine cellulaire et les programmes propres à chaque type cellulaire, même si des facteurs génétiques ou environnementaux contribuent à la modifier [14]. Le profil de méthylation renseigne donc sur la nature d’une tumeur et peut aider au diagnostic, en complément de l’analyse histopathologique. Il peut permettre de déterminer l’origine d’un cancer indifférencié, notamment en cas de métastases chez un patient sans antécédent cancérologique, et contribuer ainsi à choisir les traitements les plus adaptés [16, 19–21](→).
(→) Voir m/s n° 12, 2024, page 925
Le méthylome : méthodes et analyse
Méthodes
Une méthode d’analyse courante du profil de méthylation est basée sur le traitement de l’ADN au bisulfite. L’analyse réalisée après le traitement au bisulfite permet de distinguer les cytosines non méthylées, qui sont converties en uraciles par le bisulfite, des bases 5-méthylcytosines, qui, après séquençage, sont lues comme des cytosines et des thymines, respectivement. La technique de microarray BeadChip, telle que celle développée par la société Illumina (infinium methylationEPIC beadchip), permet un génotypage des îlots CpG avec une résolution d’une paire de bases1.
Analyse des profils de méthylation
À partir de l’état de méthylation d’un ensemble d’îlots CpG, un algorithme d’apprentissage automatique, tel que le Random Forest, permet de classer les échantillons dans des groupes aussi homogènes possibles [21]. Le résultat est un arbre décisionnel qui assigne la tumeur étudiée à un groupe de tumeurs donné. Le centre de recherche contre le cancer DKFZ (deutsches krebsforschungszentrum) d’Heidelberg, en Allemagne, a développé un nouvel algorithme d’apprentissage supervisé (forme d’intelligence artificielle) qui permet de classer les tumeurs du système nerveux central à partir des données du méthylome (Figure 1)2. La probabilité que la tumeur appartienne à un groupe donné, ou score calibré (SC, entre 0 et 1), représente le degré de confiance dans le diagnostic proposé par l’algorithme. La valeur seuil au-delà de laquelle le résultat est jugé fiable (meilleure balance entre sensibilité et spécificité) est couramment fixée à 0,9.
 |
Figure 1 Extrait de l’arborisation diagnostique selon la version 12.8 du classificateur de Heidelberg. Sont détaillés ici les gliomes diffus de l’adulte avec deux types principaux : les glioblastomes, qui se répartissent en différents sous-types (RTK1, RTK2, ... ) et les gliomes IDH-mutés qui comprennent principalement les astrocytomes de bas grade, les astrocytomes de haut grade (absence de codélétion 1p/19q) et les oligodendrogliomes (présence d’une codélétion 1p/19q). Ces différents types et sous-types tumoraux présentent des profils de méthylation distincts permettant de les classer de manière plus objective que l’analyse au microscope. |
Une carte de localisation des tumeurs du système nerveux central
En analysant des îlots CpG répartis sur l’ensemble du génome, le profil de méthylation devient pangénomique et permet de visualiser les anomalies du nombre de copies de chaque région chromosomique (gains ou pertes de segments d’ADN voire de chromosomes entiers) (Figure 2) [22]. D’un point de vue mathématique, un tel profil correspond à un espace qui possède autant de dimensions qu’il existe de loci différentiellement méthylés. Un tel espace ne peut être directement analysé visuellement ou statistiquement. Des algorithmes de réduction de dimensions, tels que le t-SNE (t-distributed stochastic neighbor embedding) ou l’UMAP (uniform manifold approximation and projection) sont donc essentiels afin de réduire le nombre de dimensions tout en restant au plus près du profil original (Figure 3).
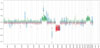 |
Figure 2 Profil pangénomique obtenu via l’analyse du méthylome. Les chromosomes sont représentés en abscisse, du chromosome 1 au chromosome 22 (de gauche à droite). Pour chaque chromosome, la ligne en pointillés représente le centromère, avec à gauche de cette ligne le bras court et à droite le bras long. En ordonnée figure l’intensité du signal de fluorescence avec, dans les valeurs positives (au-dessus de la ligne de base), les gains/amplifications et, dans les valeurs négatives, les pertes/délétions homozygotes. Cette tumeur présente un gain du chromosome 7 et une perte des chromosomes 10 et 9p, associés à une amplification du gène EGFR en 7p11 et à une délétion homozygote des gènes CDKN2A/B en 9p21. Ces anomalies de nombre sont très évocatrices d’un glioblastome IDH-non muté, particulièrement l’association gain du 7/perte du 10 et l’amplification d’EGFR. La tumeur présente par ailleurs un gain des chromosomes 19 et 20. |
 |
Figure 3 Visualisation après réduction de dimensions par t-SNE. La réduction de dimensions permet de visualiser, comme sur une carte géographique, l’appartenance d’une tumeur à un groupe de tumeurs. Chaque nuage de couleur correspond à un type ou sous-type tumoral, caractérisé par un profil de méthylation spécifique. La croix (indiquée par la flèche rouge) correspond à la tumeur étudiée. À droite, figure le code couleur avec le nom de différents types ou sous-types tumoraux présents dans la base de données. La tumeur étudiée se classe dans le groupe des glioblastomes IDH-non mutés, sous-type RTK1. L’algorithme utilisé ne peut classer correctement la tumeur étudiée que si des tumeurs de même nature (aux profils de méthylation similaires) sont présentes dans la base de données. Si le groupe est absent de la carte géographique, la tumeur se placera de façon aberrante dans un autre groupe. La liste des abréviations des diagnostics est disponible sur le site du DKFZ3. |
À partir d’un grand nombre de profils pangénomiques issus de tumeurs bien caractérisées, les algorithmes t-SNE et UMAP définissent des groupes homogènes. Ces derniers peuvent être représentés en deux ou trois dimensions comme des nuages de points indépendants.
On peut alors visualiser, comme sur une carte géographique, à quel groupe appartient une tumeur donnée (Figure 3).
La place du méthylome en neuro-oncologie
L’étude princeps qui a révolutionné la discipline
L’introduction du méthylome a été une véritable révolution pour les neuropathologistes impliqués dans le diagnostic des tumeurs du système nerveux central [23]. L’étude princeps menée par Capper et al., publiée en 2018, a porté sur 1 104 tumeurs du système nerveux central et a permis d’identifier 82 classes de tumeurs, définies chacune par un profil de méthylation distinct [24]. Parmi les 1 104 tumeurs analysées, 977 (88 %) présentaient un score calibré supérieur à 0,9 [23]. Dans 76 % des cas (838/1 104), le diagnostic intégré selon l’OMS et la classe de méthylation proposée par l’IA étaient concordants. Dans 16 % des cas (171/1 104), la tumeur a été classée par le méthylome dans un sous-groupe moléculaire que l’histopathologie seule ne pouvait pas identifier. Dans 13 % des cas (139/1 104), il existait une discordance entre le diagnostic histopathologique et la classe de méthylation. Dans 129/139 cas (soit 12 % de la cohorte totale), des analyses moléculaires complémentaires (séquençage NGS4 d’un panel plus large de gènes, séquençage des ARN ou recherche d’anomalies chromosomiques) ont permis de réviser le diagnostic en faveur de la classe de méthylation proposée. Cette révision du diagnostic a entraîné un changement de grade dans 71 % des cas (92/129), avec une augmentation et une diminution de celui-ci dans, respectivement, 41 % des cas (53/129) et 30 % des cas (39/129). 12 % des cas (127/1 104) n’ont pu être classés (score calibré inférieur à 0,9) et une réduction de dimensions par t-SNE plaçait ces cas en dehors des classes de méthylation connues, suggérant de nouveaux types tumoraux. Dix cas (moins de 1 % de la cohorte totale) n’ont pas pu être résolus et le diagnostic histopathologique a prévalu. Au total, la sensibilité et la spécificité du méthylome étaient respectivement de 0,98 et 0,99 [23].
Un outil diagnostique performant
D’autres études ont évalué la fiabilité du méthylome en confrontant le diagnostic proposé par l’IA au diagnostic intégré de l’OMS [24–29]. Dans l’étude menée par Wu et al., qui porte sur 1 258 tumeurs du système nerveux central, 694 avaient un score calibré suffisant (fixé comme supérieur à 0,84) [24]. La moitié des diagnostics proposés par l’analyse du méthylome étaient concordants avec le diagnostic intégré (53 %, 369/694). Dans 27 % des cas (187/694), un nouveau diagnostic a été retenu et dans presque 20 % des cas (137/694), le diagnostic a été affiné (proposition d’un sous-type tumoral). De plus, l’analyse du t-SNE a permis de mettre en évidence de nouveaux types tumoraux (formant des groupes distincts), non répertoriés dans la base de données du DKFZ [24].
Chez l’enfant, du fait de la grande diversité des tumeurs du système nerveux central, une analyse du méthylome est souvent nécessaire [4]. Les aspects morphologiques des tumeurs du système nerveux central pédiatriques sont variables et les diagnostics histopathologiques seuls peuvent être subjectifs. Dans une étude portant sur des tumeurs du système nerveux central de l’enfant et de l’adolescent (moins de 20 ans), 306 cas ont été classés selon le diagnostic histomoléculaire et selon le méthylome [23]. 49 % des cas (149/306) avaient un score calibré supérieur à 0,9. Pour 66 % des cas (99/149), l’analyse du méthylome a permis de confirmer et d’affiner le diagnostic. Dans ces cas, les techniques histopathologiques et de biologie moléculaire classiques étaient en défaut. Pour cinq cas, l’analyse du méthylome a permis de réviser le diagnostic histopathologique en proposant notamment de nouveaux types tumoraux désormais reconnus par l’OMS. Dans 26 % des cas (39/149), l’analyse du méthylome n’a apporté aucune information diagnostique ou pronostique supplémentaire. À l’exception de 3 cas (3/149, 2 %), l’analyse du méthylome n’a pas proposé de diagnostic erroné au vu des données histopathologiques et radiologiques [25].
Chez l’adulte, une analyse du méthylome est effectuée plus rarement. L’indication principale est une tumeur d’aspect inhabituel, difficile à classer, sans altération moléculaire clé identifiée. À noter que, selon l’OMS 2021, le diagnostic de certains types tumoraux nécessite la réalisation d’une étude du méthylome [4]. Par exemple, les épendymomes de la fosse cérébrale postérieure, rencontrés principalement chez l’adulte, ne présentent pas d’altération génétique spécifique (mutation ponctuelle ou fusion de gènes, par exemple). Leur diagnostic repose donc sur les aspects histopathologiques et un profil de méthylation concordant [4].
L’analyse du méthylome a également sa place dans l’évaluation du pronostic des tumeurs du système nerveux central. Les méningiomes se répartissent selon des profils de méthylation corrélés au risque de récidive et de transformation maligne plus pertinents que le grade proposé dans la classification de l’OMS [30, 31]5. Les médulloblastomes, tumeurs embryonnaires du petit enfant, ont une grande hétérogénéité morphologique et biologique [3]. L’analyse de leurs méthylomes a permis leur démembrement en identifiant des sous-groupes aux caractéristiques cliniques, moléculaires et pronostiques distinctes, permettant d’adapter les traitements (Figure 4) [4].
 |
Figure 4 Diagramme de Sankey. Flux de redistribution des diagnostics après analyse du méthylome (données personnelles). De gauche à droite, le diagnostic pré-méthylome et le diagnostic final intégré. L’analyse du méthylome permet, selon les cas, de confirmer le diagnostic, d’apporter une précision diagnostique (en proposant un sous-type tumoral, dont le pronostic peut être distinct) ou de réviser le diagnostic (changement de famille tumorale pouvant avoir un impact sur la prise en charge thérapeutique). Par exemple, dans la partie supérieure du schéma, certains glioblastomes IDH-non mutés ont été reclassés en gliomes diffus de haut grade de type pédiatrique (pedHGG) ou en gliomes diffus de haut grade de sous-type E ou F (HGG_E/F, nouveaux sous-types tumoraux restant à définir sur le plan histomoléculaire). La liste des abréviations des diagnostics est disponible sur le site epignostix7. |
Ainsi, l’analyse du méthylome est devenue un outil incontournable en neuro-oncologie, tant sur le plan diagnostique que pronostique, tout particulièrement chez l’enfant.
Les tumeurs du système nerveux central ne sont pas les seules à bénéficier des apports de l’analyse du méthylome : une classification des sarcomes (tumeurs malignes d’origine mésenchymateuse), développée par l’équipe de Heidelberg, ainsi qu’une classification des tumeurs de la sphère ORL ou encore de celles de la peau a aussi été établie sur la base du méthylome [32–35].
Le méthylome : le nouveau gold standard ?
Les avantages
L’analyse du méthylome est devenue un outil diagnostique, pronostique et décisionnel pour le choix des traitements [36]. Elle a permis d’identifier de nouveaux types et sous-types tumoraux, intégrés dans la classification de l’OMS 2021 [4]. Le classement de certaines tumeurs ne peut s’effectuer que par cette méthode, tandis que pour d’autres, le diagnostic histomoléculaire reste suffisant.
Contrairement à l’histopathologie, l’analyse du méthylome est indépendante du ou des observateurs. Elle peut être réalisée sur tissu congelé, ou fixé au formol et inclus en paraffine, matériel utilisé en routine pour le diagnostic microscopique des tumeurs. Cette technique génère un profil pangénomique comparable à celui obtenu par CGH (comparative genomic hybridization) array6 et permet de détecter des anomalies de nombre des chromosomes. Ce double avantage « 2 en 1 » associe diagnostic méthylomique et analyse génomique (Figure 2) [37].
L’analyse du méthylome peut également être vue comme une technique « 3 en 1 » car l’analyse d’îlots CpG isolés permet d’évaluer le niveau de méthylation des régions promotrices de gènes tels que MGMT (6-O-méthylguanine méthyltransférase)8. Cette analyse permet de prédire la réponse aux agents alkylants (chimiothérapie), tels que le témozolomide, notamment dans les glioblastomes [38].
La détermination du profil de méthylation ne nécessite pas de disposer d’un ADN intègre ou en grande quantité. C’est un examen robuste qui peut être réalisé avec un ADN fragmenté ou en faible quantité. Il est donc performant là où d’autres techniques de biologie moléculaire sont en échec.
Enfin, le méthylome ouvre la porte au diagnostic par biopsie liquide, c’est-à-dire à partir de l’ADN tumoral circulant dans les liquides biologiques, tels que le plasma et le liquide céphalo-rachidien [39, 40]. Ceci permettrait de sursoir à certaines biopsies neurochirurgicales pour porter un diagnostic.
Les limites
Le coût de la technique et sa faible disponibilité sont deux obstacles à sa généralisation. De plus, tous les centres ne disposent pas de l’équipement nécessaire.
Le diagnostic d’un type tumoral donné repose sur un faisceau d’arguments cliniques, radiologiques, microscopiques et moléculaires (données de séquençage et méthylome). L’ensemble de ces éléments doit concorder. Les diagnostics les plus justes sont ceux qui intègrent toutes les données pertinentes. L’analyse du méthylome représente un niveau d’information supplémentaire et ne suffit pas à elle seule comme outil diagnostique.
Si le méthylome peut dépasser l’œil du pathologiste, il peut être pris en défaut quand le tissu renferme peu de cellules tumorales : il peut alors rendre un diagnostic de « tissu contrôle » ou de « microenvironnement inflammatoire ». Un score calibré faible (< 0,5), en lien avec une mauvaise qualité ou quantité d’ADN, sera peu contributif [24]. Les scores calibrés faibles peuvent partiellement être résolus par la réalisation d’un t-SNE et d’un UMAP, mais cela nécessite une plateforme bio-informatique performante pour visualiser, à partir de la cartographie du DKFZ, à quel groupe appartient la tumeur.
Les tumeurs très rares, inconnues de l’algorithme, seront mal classées ou resteront inclassées. Par ailleurs, certaines tumeurs n’ont pas de classe de méthylation propre et ne peuvent donc pas être individualisées [4].
Concernant l’analyse du profil pangénomique, les anomalies structurelles de petite taille (ex : délétion homozygote d’un gène) peuvent être difficiles à détecter, notamment dans les cas où l’ADN a été extrait d’un bloc d’inclusion en paraffine (l’ADN y est dégradé par la fixation au formol), et non à partir d’un fragment tumoral congelé (où l’ADN est mieux préservé) [37]. À noter que le méthylome ne permet pas de détecter les altérations génétiques (mutations ponctuelles, fusions de gènes) pouvant faire l’objet d’une thérapie ciblée. Par ailleurs, les classes de méthylation peuvent présenter une grande hétérogénéité pronostique interindividuelle, ce qui ne permet pas toujours de prendre des décisions thérapeutiques à l’échelle du patient.
Enfin, l’analyse des données du méthylome nécessite un téléchargement des fichiers sur le site du DKFZ9 afin d’obtenir des propositions diagnostiques et un score calibré. Ce libre accès génère un double gain puisque le résultat fourni par le logiciel permet au médecin pathologiste de classer la tumeur dans une famille et, en même temps, enrichit la base de données de l’équipe d’Heidelberg, rendant les algorithmes d’IA plus performants. À ce jour, la base allemande rassemble les profils de méthylation d’environ 100 000 tumeurs du système nerveux central. Cependant, le DKFZ a mis sous embargo des données concernant de nouveaux types tumoraux.
À noter que le NIH (National Institutes of Health) aux États-Unis a également développé un algorithme disponible gratuitement avec, en plus, la possibilité de visualiser le positionnement géographique de la tumeur via un UMAP10.
Le séquençage par nanopore, une alternative à la technique « BeadChip »
Moins onéreux que la technique « Beadchip », le séquençage de l’ADN par nanopore, s’effectue en mesurant les variations d’un signal électrique généré lors du passage de l’ADN dans un nanopore, sous l’action d’une protéine motrice. Les variations du signal électrique (spécifiques à chaque base et à chaque modification épigénétique) sont décodées pour déterminer la séquence de la molécule [41] (→).
(→) Voir m/s n° 2, 2018, page 161
Puisque la 5-méthylcytosine génère une signature électrique distincte de celle des autres bases, le traitement au bisulfite de l’ADN n’est pas nécessaire. Cette technique s’est avérée aussi efficace que les puces EPIC® d’Illumina dans le diagnostic des tumeurs du système nerveux central, ainsi que pour déterminer le profil de méthylation du promoteur du gène MGMT ou détecter des anomalies chromosomiques [42]. Plus rapide que la technique « BeadChip », le séquençage par nanopore permet de classer une tumeur dans la journée (au lieu de plusieurs jours), voire pendant une intervention chirurgicale, aidant ainsi à choisir la thérapie adaptée dans les contextes urgents [43, 44]. Contrairement aux puces Illumina, le nanopore permet également de séquencer des régions d’intérêt et de détecter des mutations dans des gènes clés, tels que les gènes IDH1/2 (dans les gliomes diffus de l’adulte), ou dans le promoteur du gène TERT (telomerase reverse transcriptase) [42]. Cependant, sa résolution reste inférieure à celle de la technique « BeadChip », avec un nombre limité d’îlots CpG pouvant être analysés et une profondeur de séquençage plus faible [44]. Ses performances sont également moindres à partir d’ADN issu de tissus fixés au formol et inclus en paraffine, par rapport à des échantillons frais ou congelés. Par ailleurs, cette technique nécessite des quantités d’ADN plus importantes.
Conclusion et perspectives
L’analyse du méthylome est devenue un « examen complémentaire » incontournable en neuro-oncologie. Son avènement a révolutionné le diagnostic histomoléculaire des tumeurs du système nerveux central. Elle fait désormais partie intégrante d’un raisonnement médical regroupant à la fois les données cliniques, radiologiques, histopathologiques, génétiques et épigénétiques. Un diagnostic ne peut pas reposer uniquement sur les données du méthylome analysées par des algorithmes d’intelligence artificielle. Malgré des diagnostics plus précis, l’impact thérapeutique de l’analyse du méthylome reste limité. Les choix thérapeutiques reposent essentiellement sur les données de séquençage à grande échelle (ADN et ARN), pouvant orienter vers des thérapies ciblées.
Depuis la publication princeps de 2018, la base de données d’Heidelberg n’a fait que croître grâce au chargement, dans ce pipeline unique, de données fournies par les pathologistes du monde entier. Les fichiers soumis sont analysés et conservés, permettant d’une part la mise à jour des versions de l’algorithme, et d’autre part la découverte de nouveaux types ou sous-types tumoraux. Le système de classification du DKFZ, basé sur plus de 100 000 tumeurs, pourrait, dans quelques années, devenir une IA indépendante et être reconnu comme « expert ». Dans une médecine où seule l’obligation de moyens, et non de résultats, est en vigueur, nous serons sans doute un jour confrontés à l’obligation d’analyser le méthylome de toute tumeur du système nerveux central (voire de tout cancer).
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Infinium MethylationEPIC v2.0 Kit. https://www.illumina.com/products/by-type/microarray-kits/infinium-methylation-epic.html
Le séquençage NGS (next-generation sequencing, ou séquençage de nouvelle génération) est une technologie qui permet de séquencer l’ADN ou l’ARN de manière massivement parallèle, c’est-à-dire en lisant simultanément des millions de fragments (ndlr).
La puce d’hybridation génomique comparative, encore appelée Array comparative genomic hybridization (CGH array, array CGH, a-CGH ou aCGH) ou Chromosomal Microarray Analysis (CMA), est une technique de cytogénétique sur puces permettant d’analyser les variations du nombre de copies dans l’ADN (ndlr).
La MGMT (O6 méthyl-guanine méthyl-transférase) est une enzyme qui participe à la réparation de l’ADN et qui, par son mécanisme d’action, va supprimer les groupements alkyls formés par les chimiothérapies à base d’agents alkylants, et ainsi induire des chimio-résistances (ndlr).
Références
- Ostrom QT, Cioffi G, Waite K, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the united states in 2014-2018. Neuro Oncol 2021 ; 23 : 1–105. [Google Scholar]
- Cowppli-Bony A, Delafosse P, Lacour B, et al. Survie des atteintes de cancer en France métropolitaine 1989-2018 – Système nerveux central glioblastomes. Boulogne-Billancourt (France) : Institut national du cancer 2020, 10 p. [Google Scholar]
- Roger Stupp, Mason WP, Bent MJ van den, et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. New England Journal of Medicine 2005 ; 352 : 987–96. [CrossRef] [PubMed] [Google Scholar]
- WHO Classification of Tumours Editorial Board. Central nervous system tumours. Lyon (France) : International Agency for Research on Cancer 2021 : 508 p. [Google Scholar]
- WHO Classification of Tumours Editorial Board. Central nervous system tumours. Lyon (France) : International Agency for Research on Cancer 2007 : 309 p. [Google Scholar]
- WHO Classification of Tumours Editorial Board. Central nervous system tumours. Lyon (France) : International Agency for Research on Cancer 2016 : 408 p. [Google Scholar]
- Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology 2013 ; 38 : 23–38. [CrossRef] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009 ; 10 : 295–304. [CrossRef] [PubMed] [Google Scholar]
- Cain JA, Montibus B, Oakey RJ. Intragenic CpG islands and their impact on gene regulation. Front Cell Dev Biol 2022 ; 10 : 832348. [CrossRef] [PubMed] [Google Scholar]
- Weber M. Profils de méthylation de l’ADN dans les cellules normales et cancéreuses. Med Sci (Paris) 2008 ; 24 : 731–4. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Liu Y, Weyrich LS, Llamas B. More arrows in the ancient DNA quiver: use of paleoepigenomes and paleomicrobiomes to investigate animal adaptation to environment. Mol Biol Evol 2020 ; 37 : 307–19. [CrossRef] [PubMed] [Google Scholar]
- Pang Y-Y, Lu RJ-H, Chen P-Y. Behavioral epigenetics: perspectives based on experience-dependent epigenetic inheritance. Epigenomes 2019 ; 3 : 18. [CrossRef] [PubMed] [Google Scholar]
- Habash NW, Sehrawat TS, Shah VH, et al. Epigenetics of alcohol-related liver diseases. JHEP Rep 2022 ; 4 : 100466. [CrossRef] [PubMed] [Google Scholar]
- Loyfer N, Magenheim J, Peretz A, et al. A DNA methylation atlas of normal human cell types. Nature 2023 ; 613 : 355–64. [CrossRef] [PubMed] [Google Scholar]
- Liu C, Marioni RE, Hedman ÅK, et al. A DNA methylation biomarker of alcohol consumption. Mol Psychiatry 2018 ; 23 : 422–33. [CrossRef] [PubMed] [Google Scholar]
- Shimizu D, Taniue K, Matsui Y, et al. Pan-cancer methylome analysis for cancer diagnosis and classification of cancer cell of origin. Cancer Gene Ther 2022 ; 29 : 428–36. [CrossRef] [Google Scholar]
- Ghosh S, Yates AJ, Fruhwald MC, et al. Tissue specific DNA methylation of CpG islands in normal human adult somatic tissues distinguishes neural from non-neural tissues. Epigenetics 2010 ; 5 : 527–38. [CrossRef] [PubMed] [Google Scholar]
- Ladd-Acosta C, Pevsner J, Sabunciyan S, et al. DNA methylation signatures within the human brain. Am J Hum Genet 2007 ; 81 : 1304–15. [CrossRef] [Google Scholar]
- Möhrmann L, Werner M, Oleś M, et al. Comprehensive genomic and epigenomic analysis in cancer of unknown primary guides molecularly-informed therapies despite heterogeneity. Nat Commun 2022 ; 13 : 4485. [CrossRef] [PubMed] [Google Scholar]
- Moran S, Martínez-Cardús A, Sayols S, et al. Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol 2016 ; 17 : 1386–95. [CrossRef] [PubMed] [Google Scholar]
- Gorse M, Bianchi C, Proudhon C. Épigénétique et cancer — La méthylation dans tous ses états. Med Sci (Paris) 2024 ; 40 : 925–34. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Daenekas B, Pérez E, Boniolo F, et al. Conumee 2.0: enhanced copy-number variation analysis from DNA methylation arrays for humans and mice. Bioinformatics 2024 ; 40 : btae029. [CrossRef] [Google Scholar]
- Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature 2018 ; 555 : 469–74. [CrossRef] [PubMed] [Google Scholar]
- Wu Z, Abdullaev Z, Pratt D, et al. Impact of the methylation classifier and ancillary methods on CNS tumor diagnostics. Neuro Oncol 2021 ; 24 : 571–81. [Google Scholar]
- Pickles JC, Fairchild AR, Stone TJ, et al. DNA methylation-based profiling for paediatric CNS tumour diagnosis and treatment: a population-based study. Lancet Child Adolesc Health 2020 ; 4 : 121–30. [CrossRef] [PubMed] [Google Scholar]
- Pages M, Uro-Coste E, Colin C, et al. The implementation of dna methylation profiling into a multistep diagnostic process in pediatric neuropathology: a 2-year real-world experience by the french neuropathology network. Cancers (Basel) 2021 ; 13 : 1377. [CrossRef] [Google Scholar]
- Sturm D, Capper D, Andreiuolo F, et al. Multiomic neuropathology improves diagnostic accuracy in pediatric neuro-oncology. Nat Med 2023 ; 29 : 917–26. [CrossRef] [PubMed] [Google Scholar]
- Capper D, Stichel D, Sahm F, et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol 2018 ; 136 : 181–210. [CrossRef] [Google Scholar]
- Jaunmuktane Z, Capper D, Jones DTW, et al. Methylation array profiling of adult brain tumours: diagnostic outcomes in a large, single centre. Acta Neuropathol Commun 2019 ; 7 : 24. [CrossRef] [Google Scholar]
- Choudhury A, Magill ST, Eaton CD, et al. Meningioma DNA methylation groups identify biological drivers and therapeutic vulnerabilities. Nat Genet 2022 ; 54 : 649–59. [CrossRef] [PubMed] [Google Scholar]
- Sahm F, Schrimpf D, Stichel D, et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol 2017 ; 18 : 682–94. [CrossRef] [PubMed] [Google Scholar]
- Koelsche C, Schrimpf D, Stichel D, et al. Sarcoma classification by DNA methylation profiling. Nat Commun 2021 ; 12 : 498. [CrossRef] [PubMed] [Google Scholar]
- Jurmeister P, Glöß S, Roller R, et al. DNA methylation-based classification of sinonasal tumors. Nat Commun 2022 ; 13 : 7148. [CrossRef] [PubMed] [Google Scholar]
- Koelsche C, Deimling A von. Methylation classifiers: brain tumors, sarcomas, and what’s next. Genes Chromosomes Cancer 2022 ; 61 : 346–55. [CrossRef] [PubMed] [Google Scholar]
- Rodríguez-Paredes M, Bormann F, Raddatz G, et al. Methylation profiling identifies two subclasses of squamous cell carcinoma related to distinct cells of origin. Nat Commun 2018 ; 9 : 577. [CrossRef] [PubMed] [Google Scholar]
- Khalighi S, Reddy K, Midya A, et al. Artificial intelligence in neuro-oncology: advances and challenges in brain tumor diagnosis, prognosis, and precision treatment. NPJ Precis Oncol 2024 ; 8 : 80. [CrossRef] [PubMed] [Google Scholar]
- Kilaru V, Knight AK, Katrinli S, et al. Critical evaluation of copy number variant calling methods using DNA methylation. Genet Epidemiol 2020 ; 44 : 148. [CrossRef] [PubMed] [Google Scholar]
- Della Monica R, Cuomo M, Buonaiuto M, et al. MGMT and whole-genome DNA methylation impacts on diagnosis, prognosis and therapy of glioblastoma multiforme. Int J Mol Sci 2022 ; 23 : 7148. [CrossRef] [PubMed] [Google Scholar]
- Sabedot TS, Malta TM, Snyder J, et al. A serum-based DNA methylation assay provides accurate detection of glioma. Neuro Oncol 2021 ; 23 : 1494–508. [CrossRef] [PubMed] [Google Scholar]
- An S, McCortney K, Walshon J, et al. Methylation profiling of plasma cell-free DNA in pediatric brain tumor patients. Acta Neuropathol 2024 ; 148 : 29. [CrossRef] [Google Scholar]
- Montel F. Séquençage de l’ADN par nanopores — Résultats et perspectives. Med Sci (Paris) 2018 ; 34 : 161–5. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Patel A, Dogan H, Payne A, et al. Rapid-CNS2: rapid comprehensive adaptive nanopore-sequencing of CNS tumors, a proof-of-concept study. Acta Neuropathol 2022 ; 143 : 609–12. [CrossRef] [Google Scholar]
- Kuschel LP, Hench J, Frank S, et al. Robust methylation-based classification of brain tumours using nanopore sequencing. Neuropathol Appl Neurobiol 2023 ; 49 : e12856. [CrossRef] [PubMed] [Google Scholar]
- Vermeulen C, Pagès-Gallego M, Kester L, et al. Ultra-fast deep-learned CNS tumour classification during surgery. Nature 2023 ; 622 : 842–9. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1 Extrait de l’arborisation diagnostique selon la version 12.8 du classificateur de Heidelberg. Sont détaillés ici les gliomes diffus de l’adulte avec deux types principaux : les glioblastomes, qui se répartissent en différents sous-types (RTK1, RTK2, ... ) et les gliomes IDH-mutés qui comprennent principalement les astrocytomes de bas grade, les astrocytomes de haut grade (absence de codélétion 1p/19q) et les oligodendrogliomes (présence d’une codélétion 1p/19q). Ces différents types et sous-types tumoraux présentent des profils de méthylation distincts permettant de les classer de manière plus objective que l’analyse au microscope. |
| Dans le texte | |
|
Figure 2 Profil pangénomique obtenu via l’analyse du méthylome. Les chromosomes sont représentés en abscisse, du chromosome 1 au chromosome 22 (de gauche à droite). Pour chaque chromosome, la ligne en pointillés représente le centromère, avec à gauche de cette ligne le bras court et à droite le bras long. En ordonnée figure l’intensité du signal de fluorescence avec, dans les valeurs positives (au-dessus de la ligne de base), les gains/amplifications et, dans les valeurs négatives, les pertes/délétions homozygotes. Cette tumeur présente un gain du chromosome 7 et une perte des chromosomes 10 et 9p, associés à une amplification du gène EGFR en 7p11 et à une délétion homozygote des gènes CDKN2A/B en 9p21. Ces anomalies de nombre sont très évocatrices d’un glioblastome IDH-non muté, particulièrement l’association gain du 7/perte du 10 et l’amplification d’EGFR. La tumeur présente par ailleurs un gain des chromosomes 19 et 20. |
| Dans le texte | |
|
Figure 3 Visualisation après réduction de dimensions par t-SNE. La réduction de dimensions permet de visualiser, comme sur une carte géographique, l’appartenance d’une tumeur à un groupe de tumeurs. Chaque nuage de couleur correspond à un type ou sous-type tumoral, caractérisé par un profil de méthylation spécifique. La croix (indiquée par la flèche rouge) correspond à la tumeur étudiée. À droite, figure le code couleur avec le nom de différents types ou sous-types tumoraux présents dans la base de données. La tumeur étudiée se classe dans le groupe des glioblastomes IDH-non mutés, sous-type RTK1. L’algorithme utilisé ne peut classer correctement la tumeur étudiée que si des tumeurs de même nature (aux profils de méthylation similaires) sont présentes dans la base de données. Si le groupe est absent de la carte géographique, la tumeur se placera de façon aberrante dans un autre groupe. La liste des abréviations des diagnostics est disponible sur le site du DKFZ3. |
| Dans le texte | |
|
Figure 4 Diagramme de Sankey. Flux de redistribution des diagnostics après analyse du méthylome (données personnelles). De gauche à droite, le diagnostic pré-méthylome et le diagnostic final intégré. L’analyse du méthylome permet, selon les cas, de confirmer le diagnostic, d’apporter une précision diagnostique (en proposant un sous-type tumoral, dont le pronostic peut être distinct) ou de réviser le diagnostic (changement de famille tumorale pouvant avoir un impact sur la prise en charge thérapeutique). Par exemple, dans la partie supérieure du schéma, certains glioblastomes IDH-non mutés ont été reclassés en gliomes diffus de haut grade de type pédiatrique (pedHGG) ou en gliomes diffus de haut grade de sous-type E ou F (HGG_E/F, nouveaux sous-types tumoraux restant à définir sur le plan histomoléculaire). La liste des abréviations des diagnostics est disponible sur le site epignostix7. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.