")
")
| Issue |
Med Sci (Paris)
Volume 41, Number 11, Novembre 2025
|
|
|---|---|---|
| Page(s) | 877 - 886 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2025187 | |
| Published online | 12 December 2025 | |
Sauvetage génétique somatique dans les maladies hématopoïétiques à hérédité mendélienne
Du darwinisme cellulaire à la thérapie génique naturelle
Somatic genetic rescue in mendelian hematopoietic diseases: from cellular darwinism to natural gene therapy
1
Laboratoire de dynamique du génome dans les maladies humaines, Équipe labellisée Ligue contre le Cancer “Ligue 2023”, Inserm UMR1163, Imagine Institute, Paris, France
2
Université Paris Saclay, France
3
Université Paris cité, Imagine Institute, Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
**
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
L’accumulation de mutations spontanées, dites somatiques, au cours du temps est un phénomène naturel qui se produit dans tous les tissus des individus sains. Ce phénomène contribue à la dégénérescence progressive des systèmes biologiques associée au vieillissement et au développement de cancers. Cependant, dans le contexte de maladies mendéliennes (c’est-à-dire causées par des mutations génétiques germinales), les mutations somatiques peuvent parfois contrer l’effet délétère du défaut germinal et conférer un avantage sélectif aux cellules. Ce phénomène, nommé « sauvetage génétique somatique » (somatic genetic rescue) peut avoir des conséquences importantes sur le diagnostic et l’évolution de la maladie.
Abstract
Spontaneous (somatic) mutations accumulate naturally over time in all tissues of healthy individuals. This phenomenon contributes to the progressive degeneration of biological systems associated with aging and the development of cancers. However, in the context of mendelian diseases, i.e. those caused by germline monogenic mutations, spontaneous somatic mutations can sometimes counterbalance the deleterious effect of the germline defect conferring a selective advantage to cells. This phenomenon, known as somatic genetic rescue, can have important implications for the diagnosis and progression of the disease.
© 2025 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (© Patrick Revy)
La génération de mutations somatiques constitue un phénomène naturel qui s’opère chez tous les êtres vivants, y compris l’être humain. Une mutation somatique est définie comme toute modification génétique survenant dans une cellule après la fécondation et pouvant être générée tout au long de la vie d’un individu. Ces mutations peuvent survenir de manière spontanée, en raison d’anomalies dans les mécanismes de réparation ou de réplication de l’ADN, ou encore lors de processus intracellulaires générant des radicaux libres mutagènes. Les mutations somatiques peuvent également être induites par des stress exogènes tels que l’inflammation, l’exposition à des radiations ou à des produits chimiques [1-4]. Les progrès récents des techniques de séquençage ont permis de mettre en évidence que chaque individu accumule, au cours de sa vie, des mutations somatiques dans tous les tissus. La fréquence et la nature des mutations somatiques sont très similaires dans la plupart des tissus, à l’exception des cellules germinales qui présentent une fréquence de mutations somatiques réduite [5]. En outre, un taux similaire de mutations somatiques est mesuré dans les neurones post-mitotiques et dans des cellules qui prolifèrent, telles que les cellules du système hématopoïétique, suggérant que la mutagénèse somatique est majoritairement indépendante de la division cellulaire et de la nature du tissu. Il est maintenant établi que l’accumulation de mutations somatiques au cours du temps chez des individus sains contribue à la dégénérescence progressive des tissus, à l’apparition de maladies associées au vieillissement et à la cancérogenèse [5-10]. Par conséquent, l’acquisition de mutations somatiques est considérée comme un processus délétère pour les individus.
Cependant, dans le contexte des maladies génétiques mendéliennes (c’est-à-dire causées par des mutations pathogènes portées par les cellules germinales), l’acquisition d’une mutation somatique peut parfois atténuer ou neutraliser l’impact délétère de la mutation germinale. Ce phénomène, nommé « sauvetage génétique somatique » (ou SGR pour «somatic genetic rescue») confère un avantage sélectif à la cellule portant la mutation somatique. Ainsi, dans des cellules capables de se diviser, le SGR pourra, par exemple, entraîner une expansion clonale des cellules somatiquement modifiées (Figure 1). Dans le contexte de maladies génétiques mendéliennes, le phénomène de SGR peut être considéré comme une illustration du darwinisme cellulaire [11]. Jusqu’il y a peu, les évènements de SGR étaient considérés comme rares, voire exceptionnels. Cependant l’avènement de techniques de séquençage de nouvelle génération, beaucoup plus sensibles et fiables, ont permis au cours de la dernière décennie de révéler que le phénomène de SGR est beaucoup plus fréquent qu’anticipé.
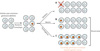 |
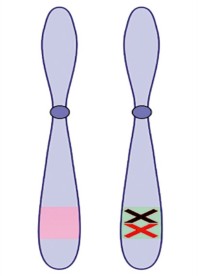
Figure 1 Principe du sauvetage génétique somatique. Dans le contexte d’une maladie à hérédité mendélienne (avec cellules porteuses d’une mutation germinale délétère), une mutation somatique peut avoir plusieurs effets : A. elle peut être délétère et conduire à la disparition de la cellule par apoptose ou immunosurveillance ; B. elle peut être neutre, et les cellules porteuses de la mutation somatique se comporteront comme les cellules non modifiées ; C. elle peut contrer l’effet délétère de la mutation germinale et conférer un avantage sélectif aux cellules somatiquement modifiées et provoquer une expansion clonale. Ce dernier cas correspond au mécanisme de sauvetage génétique somatique. Les cas B. et C. génèrent du mosaïcisme cellulaire. Les cellules portant une mutation somatique sont représentées avec un noyau orange. La membrane lobée représente les cellules dont la fonction est altérée par la mutation germinale |
Bien que le SGR puisse en théorie survenir dans toutes les maladies mendéliennes, il a été particulièrement décrit dans les maladies génétiques affectant le système hématopoïétique. En effet, les cellules sanguines sont plus facilement accessibles que d’autres tissus et leur capacité proliférative permet de détecter aisément des expansions clonales de cellules portant des SGR (Figure 1).
Dans cette revue, nous décrirons les principales caractéristiques génétiques du SGR que nous illustrerons par des exemples rencontrés dans les maladies mendéliennes hématopoïétiques. Nous discuterons également de l’impact potentiel du SGR sur le diagnostic, l’évolution clinique des malades, et l’identification de nouvelles pistes thérapeutiques.
Maladies hématopoïétiques héréditaires et sauvetage génétique somatique : généralités
Les maladies hématopoïétiques à hérédité mendélienne sont causées par des mutations germinales qui provoquent un défaut de production et/ou de fonction d’au moins une population cellulaire sanguine. Dans ces maladies, toute mutation somatique qui, en contrant spécifiquement l’effet délétère de la mutation germinale, confère une meilleure capacité d’expansion, de différenciation, de migration ou de survie par rapport aux cellules dépourvues de modification somatique sera considérée comme un SGR. L’avantage sélectif induit par le SGR provoque une expansion clonale des cellules portant la mutation somatique, générant ainsi un mosaïcisme dans le sang (cf section 4) [12].
Il est important de noter que les mutations somatiques représentant des SGR chez des patients atteints de maladies hématopoïétiques mendéliennes sont à distinguer des mutations somatiques à l’origine d’expansions clonales observées chez la plupart des individus sains âgés, un phénomène nommé « hématopoïèse clonale à potentiel indéterminé » (clonal hematopoiesis of indeterminate potential ; CHIP en anglais) [13]. En effet, la CHIP consiste en l’expansion clonale de cellules sanguines portant des mutations somatiques dans des gènes impliqués dans des hémopathies myéloïdes ou lymphoïdes, sans toutefois nécessairement provoquer de cancers chez ces individus.
À ce jour, des phénomènes de SGR ont été rapportés dans plus de quarante maladies mendéliennes hématopoïétiques différentes [12, 14]. On peut distinguer deux types de SGR : les SGR directs, correspondant à des mutations somatiques qui modifient le gène porteur de la mutation germinale et les SGR indirects, correspondant à des mutations somatiques qui touchent un autre gène que celui affecté par la mutation germinale, mais impliqué dans le processus biologique altéré par celle-ci.
Les mécanismes du sauvetage génétique somatique direct
Les mutations ponctuelles
Le SGR direct peut être causé par des mutations somatiques ponctuelles (modifiant une seule paire de bases). Des cas rares de SGR consistent en un remplacement spontané de la mutation transmise par la cellule germinale, par sa version sauvage, phénomène appelé réversion ou « back-mutation » (Figure 2A, Figure 3B). Ce type de SGR a été observé, par exemple, chez un patient atteint d’une insuffisance de la moelle osseuse causée par une mutation germinale homozygote dans le gène MYSM1 (Myb-Like, SWIRM and MPN domain-containing protein 1) qui code une histone déubiquitinase et pour lequel une réversion somatique dans une cellule souche hématopoïétique multipotente a permis de restaurer un allèle sauvage pour MYSM1 et de conférer un avantage sélectif aux cellules somatiquement modifiées [15]. Une mutation somatique ponctuelle sur le site de la mutation germinale peut aussi créer une substitution plutôt qu’un retour à la séquence sauvage et entraîner un SGR direct dans le cas où l’acide aminé codé par cette substitution est moins délétère que celui issu de la mutation germinale initiale (Figure 2B). Un exemple de ce type de SGR a été décrit dans une famille dont trois patients portaient une mutation germinale hétérozygote dans le gène GATA2 (GATA binding protein 2) codant un facteur de transcription nécessaire au développement des cellules souches hématopoïétiques. La mutation germinale correspond à un codon stop qui tronque la protéine GATA2 et cause une insuffisance de la moelle osseuse et un syndrome myélodysplasique. Chez un individu de cette famille, une mutation somatique ponctuelle corrigeant la mutation germinale survenant dans une cellule souche hématopoïétique a permis de substituer le codon délétère par un codon synonyme du codon fonctionnel sauvage et de reconstituer un système hématopoïétique fonctionnel [16].
 |
Figure 2 Les différentes possibilités de mutations ponctuelles associées aux SGR. |
 |
Figure 3 Exemples d’événements génétiques somatiques associés à des SGR directs. Dans le contexte d’une maladie à hérédité mendélienne causée par une mutation germinale hétérozygote avec effet dominant négatif (A.), un SGR direct peut survenir par : B. mutation ponctuelle qui remplace la mutation germinale par la séquence sauvage (réversion) ; C. mutation ponctuelle non-sens qui élimine l’expression du gène portant la mutation germinale ; D. conversion génique qui copie une partie du chromosome sauvage à la place du gène portant la mutation germinale (disomie uniparentale); E. perte du chromosome (monosomie) contenant le gène portant la mutation germinale; F. délétion interstitielle éliminant une portion du chromosome incluant le gène portant la mutation germinale. |
Un SGR direct peut également résulter d’une mutation somatique sur le même gène que la mutation germinale mais à un endroit différent. Dans le cas où la mutation germinale est située dans une séquence codante et entraîne une mutation non-sens ou faux sens délétère, un SGR sur un autre nucléotide du codon portant la mutation germinale (Figure 2C) peut donner lieu à la synthèse d’un autre acide aminé moins délétère et conférer un avantage sélectif.
Si la variation génétique germinale crée une mutation gain de fonction, une mutation ponctuelle perte de fonction ou faux sens touchant un autre codon éloigné de la mutation germinale sur le même allèle (en cis) peut contrer l’effet pathogène du variant (Figure 2C, Figure 3C). Par exemple, ces mutations de second site sont fréquemment détectées dans les cellules sanguines de patients souffrants de syndromes SAMD9/SAMD9L (sterile alpha motif domain–containing protein 9 / SAMD9 -like), aussi appelés respectivement, syndrome MIRAGE et ataxie/ pancytopénie, caractérisés par une insuffisance médullaire et causés par des mutations germinales hétérozygotes gain de fonction du gène SAMD9 et SAMD9L [17]. Ces gènes, tous deux localisés sur le chromosome 7, ont pour activité principale de réduire la synthèse protéique et la croissance cellulaire, en particulier dans les cellules du système hématopoïétique. De nombreux patients atteints du syndrome SAMD9 ou SAMD9L présentent des cellules sanguines portant des SGR caractérisés par des mutations ponctuelles somatiques non-sens dans le gène portant la mutation germinale. Ces événements somatiques ont pour conséquence d’annuler l’effet gain de fonction provoqué par la mutation germinale en éliminant l’expression de la protéine mutée et en restaurant une croissance cellulaire normale (Figure 3C) [17-21].
Des mutations somatiques dans les régions non codantes telles que les régions promotrices, bien qu’éloignées des mutations germinales, peuvent constituer des SGR. Ce phénomène a été observé dans des téloméropathies, un groupe de maladies génétiques rares et hétérogènes causées par des mutations germinales dans des gènes participant à la protection et à la régulation de la taille de l’extrémité des chromosomes : les télomères [22]. Les téloméropathies sont caractérisées par des signes cliniques associés à un vieillissement accéléré qui peuvent se manifester par une dyskératose congénitale [23] (→), des défauts hématologiques (insuffisance médullaire) ou une fibrose pulmonaire [24] (→).
(→) Voir m/s n° 4, 2008, page 390
(→) Voir m/s n° 6-7, 2022, page 579
Certaines téloméropathies sont causées par des mutations hétérozygotes germinales du gène TERT (telomerase reverse transcriptase), entraînant un défaut d’activité de cette protéine et un raccourcissement accéléré des télomères [22]. La protéine TERT est la sous-unité catalytique du complexe télomérase impliquée dans la régulation de la longueur des télomères. Des mutations somatiques ponctuelles dans les cellules sanguines de patients présentant une déficience en TERT ont été détectées à trois positions spécifiques dans le promoteur de TERT. Ces dernières ont pour particularité de créer des sites de fixation de facteurs de transcription qui augmentent l’expression de TERT et compensent la réduction de la taille des télomères [14, 25-28]. De façon intéressante, dans ce cas particulier, et contrairement aux SGR identifiés dans les syndromes SAMD9/SAMD9L, les SGR du promoteur TERT se localisent préférentiellement sur l’allèle ne portant pas la mutation germinale (en trans) [63](→).
(→) Voir m/s n° 6-7, 2024, page 555
Cela a pour conséquence d’augmenter l’expression de la forme sauvage de TERT et non de celle portant la mutation germinale et, par conséquent, de conférer un avantage aux cellules en améliorant la maintenance de la longueur des télomères.
Les courtes insertions ou délétions
Un autre type de SGR direct est associé à un décalage du cadre de lecture provoqué par de courtes insertions ou délétions (indels) somatiques. Ainsi, dans certaines maladies causées par un indel germinal décalant le cadre de lecture, de courts indels somatiques ont permis de restaurer le cadre de lecture sauvage du gène [29]. Au contraire, un indel somatique peut, en décalant le cadre de lecture, générer un allèle nul et ainsi contrer l’effet délétère d’une mutation germinale dominante. Ce type de SGR a été décrit chez plusieurs patients atteints des syndromes SAMD9/SAMD9L, ainsi que chez des patients présentant des fibroses pulmonaires causées par des mutations germinales hétérozygotes dominantes du gène TINF2 (TERF1-interacting nuclear factor 2), codant la protéine TIN2, un facteur participant au recrutement de la télomérase aux télomères [14, 17, 21, 30].
La création de SGR par indel peut parfois être favorisée par la nature de la séquence du gène portant la mutation germinale. Ceci a été illustré chez un patient atteint du syndrome de Wiskott Aldrich, une maladie caractérisée par des défauts hématologiques et un déficit immunitaire, causés par une insertion germinale de six paires de bases dans le gène WASP (Wiskott-Aldrich syndrome protein), un facteur participant à l’hématopoïèse. Des séquences répétées contenues dans le gène WASP ont favorisé le glissement de l’ADN polymérase lors de la réplication, ce qui a provoqué une mutation somatique caractérisée par un décalage du cadre de lecture et par une délétion de six paires de bases restaurant ainsi l’allèle sauvage de WASP [31].
La recombinaison intragénique et la conversion génique
La recombinaison intragénique mitotique correspond à l’échange de matériel génétique provenant de chromosomes homologues issus des deux parents. Dans les maladies autosomales récessives causées par des mutations germinales hétérozygotes composites, c’est-à-dire deux variants différents en trans, localisées dans le même gène mais éloignés l’un de l’autre, la recombinaison intragénique mitotique peut regrouper les deux mutations sur un même allèle et ainsi générer un allèle sauvage. Dans certaines maladies mendéliennes, ce type de SGR par recombinaison intragénique est favorisé par le défaut moléculaire causé par la mutation germinale. Par exemple, le syndrome de Bloom, maladie principalement caractérisée par un retard de croissance et une prédisposition très élevée à développer des cancers, est causé par des mutations bialléliques affectant le gène BLM (Bloom Syndrome RecQlike helicase), un facteur régulant la recombinaison homologue. Un défaut de la protéine BLM provoque une augmentation du taux de recombinaison entre chromosomes et, par conséquent, favorise l’induction de SGR suite à une recombinaison intragénique [29, 32].
Le mécanisme de conversion génique consiste en le remplacement d’une région chromosomique par une copie de la même région issue du chromosome de l’autre parent (Figure 3D). La présence, dans une cellule, de deux chromosomes homologues (ou d’une partie de chromosome) provenant d’un seul des parents est appelée disomie uniparentale (uniparental disomy ou UPD). Dans le cas d’une maladie autosomique dominante, un mécanisme de conversion génique remplace la région portant la mutation germinale par une copie du chromosome sauvage de l’autre parent, ce qui permet donc la restauration de deux copies sauvages du gène. Ce phénomène a été récemment décrit chez plusieurs patients portant une mutation germinale hétérozygote pour le facteur de transcription hématopoïétique MECOM (MDS1 and EVI1 complex locus), associée à un syndrome d’insuffisance médullaire, et une disomie uniparentale de la région du chromosome 3q comprenant MECOM. Ceci a permis de restaurer deux allèles sauvages chez les patients [33]. La disomie uniparentale est également un SGR fréquemment détecté chez les patients atteints des syndromes liés aux gènes SAMD9/SAMD9L [17, 34].
Il semblerait que les mécanismes générant des disomies uniparentales représentent les types de SGR les plus fréquemment détectés dans les maladies hématopoïétiques mendéliennes [12, 35-38].
Les altérations de la structure des chromosomes
Des remaniements chromosomiques plus importants tels que la perte partielle (délétion chromosomique) ou complète du chromosome (monosomie, (Figure 3E) portant la mutation germinale peuvent éliminer l’effet délétère de cette mutation. Des délétions d’une partie interne des chromosomes (délétions interstitielles) peuvent également conduire à l’élimination du gène porteur de la mutation germinale (Figure 3F). De tels mécanismes ont été observés dans les syndromes liés aux gènes SAMD9/SAMD9L, pour lesquels l’adaptation par aneuploïdie ou délétion interstitielle élimine le chromosome porteur de la mutation germinale et laisse une seule copie sauvage du gène SAMD9 ou SAMD9L [17, 39]. Enfin, l’élimination d’une mutation germinale peut résulter d’un phénomène de chromothripsis1, consistant en des remaniements massifs d’un ou de plusieurs chromosomes et provoquant de larges insertions, duplications et délétions [40] (→).
(→) Voir m/s n° 3, 2014, page 266
Ce phénomène a été observé chez un patient atteint du syndrome WHIM2 causé par une mutation gain de fonction de CXCR4 (C-X-C motif chemokine receptor 4).
Un événement de chromothripsis a entraîné la perte d’une région de 35 mégabases contenant la version mutée du gène et une expansion des cellules ayant subi cette modification génétique somatique complexe [41].
Le sauvetage génétique somatique indirect
Contrairement aux exemples précédents, les événements de SGR indirects correspondent à des modifications génétiques somatiques qui n’affectent pas le gène portant la mutation germinale, mais touchent d’autres gènes impliqués dans la voie affectée par le défaut germinal. L’identification des SGR indirects est plus difficile, car le gène somatiquement modifié n’est pas celui qui porte la mutation germinale. Jusqu’à présent, des SGR indirects n’ont été décrits que dans un nombre limité de maladies.
Un exemple prototypique de SGR indirect a été rapporté dans le syndrome de Shwachman-Diamond. Ce syndrome, causé majoritairement par des mutations bialléliques germinales dans le gène SBDS (Shwachman-Bodian-Diamond syndrome protein), est une ribosomopathie principalement caractérisée par une insuffisance pancréatique exocrine et des anomalies hématologiques. SBDS est une protéine impliquée dans l’éviction du facteur eIF6 (eukaryotic translation initiation factor 6) de la sousunité ribosomale pré-60S. Le retrait d’eIF6 de la sousunité pré-60S est nécessaire pour permettre à cette dernière de s’associer à la sous-unité pré-40S, afin de former un ribosome mature qui garantit la synthèse protéique (Figure 4). Ainsi, des mutations germinales dans SBDS perturbent la production des ribosomes fonctionnels en raison d’une trop grande quantité de eIF6 restant liés aux sous-unités pré-60S [42] (Figure 4). Plusieurs types de mutations somatiques affectant le gène EIF6 ont été détectées dans des cellules de la moelle osseuse et du sang périphérique de plusieurs patients atteints du syndrome de Shwachman-Diamond. Le premier type de SGR identifié était constitué de délétions chromosomiques interstitielles comprenant la région portant le gène EIF6 [43-45]. Ces observations suggéraient que la perte d’une copie du gène EIF6 (i.e. une haploinsuffisance), en diminuant l’effet de saturation d’eIF6 sur les sous-unités pré-60S pouvait donner un avantage prolifératif aux cellules modifiées (Figure 4). Plus récemment, de nombreuses mutations somatiques ponctuelles non-sens ou déstabilisant l’expression ou la fonction d’eIF6 ont été détectées dans les cellules sanguines de patients atteints du syndrome de Shwachman-Diamond, démontrant que des événements somatiques perturbant la quantité ou la fonction d’eIF6 représentaient des SGR fréquents dans le contexte de ce syndrome [4, 46, 47].
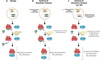 |
Figure 4 Exemple de SGR indirect dans le contexte du syndrome de Shwachman-Diamond. A. SBDS est une protéine qui retire le facteur eIF6 de la sous-unité ribosomale pré-60S afin de permettre son interaction avec la sous-unité pré-40S pour former un ribosome fonctionnel. B. Des mutations germinales délétères de SBDS affectent le mécanisme d’éviction d’eIF6, la formation de ribosome et causent le syndrome de Shwachman-Diamond. C. une mutation somatique dans le gène EIF6 (ici est représentée une délétion interstitielle) qui affecte l’expression ou la fonction anti-associative d’eIF6 représente un SGR en restaurant la formation de ribosomes fonctionnels. |
Le phénomène de SGR indirect semble également être fréquemment observé dans les téloméropathies mendéliennes [12]. Dans ce contexte, les SGR indirects affectent des facteurs qui régulent l’activité de la télomérase. Ainsi, des mutations somatiques perte de fonction dans POT1 (protection of telomeres protein 1), un facteur inhibant l’activité de la télomérase, peuvent s’avérer bénéfiques en stimulant l’activité de la télomérase et ainsi limiter le raccourcissement accéléré des télomères [14, 22, 48, 49]. De plus, les mutations somatiques résultant d’un SGR direct et affectant le promoteur du gène TERT peuvent aussi représenter des SGR indirects dans les téloméropathies causées par des mutations germinales affectant des gènes autres que le gène TERT [14, 25-28].
Des mutations somatiques dans d’autres gènes tels que PPM1D (protein phosphatase, Mg2+/Mn2+ pependent 1D), U2AF1 (U2 small nuclear RNA auxiliary factor 1), et ATM (ataxia telangiectasia mutated) ont également été identifiées chez plusieurs patients atteints de téloméropathies et sont présumées représenter des SGR [14, 50]. Cependant, pour ces gènes, à ce jour, il n’a pas été précisé comment la mutation somatique pouvait provoquer un effet bénéfique spécifiquement sur les télomères. De façon très intéressante, chez les patients atteints de téloméropathies, plusieurs mutations correspondant à des SGR représentent des mutations pro-oncogéniques. En effet, dans les téloméropathies, ces mutations contrecarrent la réduction des télomères causée par les mutations germinales et sont par conséquent protectrices. En revanche, dans le cadre du cancer, ces mêmes mutations provoquent une activation inadéquate de la télomérase et participent à la progression tumorale et à l’immortalisation cellulaire [14].
Sauvetage génétique somatique et mosaïcisme
L’avantage sélectif conféré par le SGR aux cellules somatiquement mutées peut promouvoir leur expansion et ainsi créer un mosaïcisme cellulaire (Figure 1C). La nature et l’étendue du mosaïcisme somatique généré par le SGR sont déterminées par plusieurs paramètres, parmi lesquels la nature de la cellule qui acquiert le SGR et sa capacité proliférative. En effet, un SGR présent dans des cellules très différenciées et faiblement proliférantes sera associé à un petit nombre de lignées portant le SGR. Au contraire, si le SGR survient dans une cellule souche hématopoïétique ou un progéniteur multipotent capable de produire les différentes lignées du système hématopoïétique et si l’avantage sélectif est fort, toutes les cellules du sang pourront porter la mutation. Ceci a été observé dans quelques cas où le SGR, probablement acquis dans une seule cellule souche hématopoïétique, a permis de reconstituer un système hématopoïétique dans lequel toutes les cellules portent la mutation somatique [15, 16, 41, 51]. Dans d’autres cas, l’avantage conféré par le SGR peut être spécifique à un type cellulaire particulier ou se produire dans une cellule déjà différenciée. Par exemple, dans le cas d’une mutation germinale affectant un gène nécessaire à l’activation des lymphocytes T, un SGR conférera un avantage prolifératif uniquement dans ces cellules. Ainsi, les SGR ne s’accompagnent pas nécessairement d’une expansion clonale importante ni d’un mosaïcisme massif [12].
Fréquence du sauvetage génétique somatique
Jusqu’à peu, il était présumé que le SGR était un évènement extrêmement rare. Or, l’analyse génétique de maladies hématopoïétiques mendéliennes à l’aide de nouvelles techniques de séquençage sensibles a permis de révéler que les SGR sont fréquemment détectés dans un nombre croissant de maladies [12, 14, 16, 17, 21, 22, 35, 46, 47]. À titre d’exemple, des études récentes ont révélé que des clones sanguins porteurs de SGR affectant le gène EIF6 étaient présents chez plus de 60 % des patients atteints du syndrome de Shwachman-Diamond [46, 47]. Par ailleurs, ces études ont révélé que plusieurs clones sanguins portant des SGR différents touchant EIF6 pouvaient être présents chez un même patient. De façon similaire, une étude récente réalisée sur une large cohorte de patients atteints de téloméropathies a révélé que 46 % d’entre eux présentaient des SGR [14]. De même, l’analyse de nombreux patients atteints des syndromes liés aux gènes SAMD9L/SAMD9 a révélé que 61 % d’entre eux présentaient des clones sanguins porteurs de SGR [17]. Globalement, ces observations semblent indiquer que l’existence de SGR dans un contexte de maladie hématopoïétique mendélienne est relativement fréquente.
Bénéfices thérapeutiques du sauvetage génétique somatique
Bien que la détection de clones circulants porteurs de SGR suggère que le sauvetage offre un avantage sélectif au niveau cellulaire, cet effet ne se traduit pas nécessairement par des améliorations cliniques détectables. L’effet clinique bénéfique d’un SGR est très dépendant de son degré de mosaïcisme. Ainsi, chez un grand nombre de patients présentant des clones cellulaires sanguins portant des SGR avec un faible mosaïcisme, on n’observe pas d’amélioration clinique évidente. Cependant, il a été proposé que dans plusieurs maladies hématopoïétiques (syndrome de Shwachman-Diamond, téloméropathies, et syndromes liés aux gènes SAMD9/SAMD9L), les SGR, bien que n’améliorant pas les paramètres hématologiques de façon évidente, pourraient stabiliser les défauts hématologiques voire exercer une fonction protectrice en prévenant le développement de syndromes myélodysplasiques et de cancers [4, 14, 17, 25, 35, 47, 48]. Des études longitudinales plus poussées sont nécessaires pour préciser l’évolution clinique des patients porteurs de SGR sur des temps longs.
Néanmoins, il existe des cas où le SGR apparaît dans une cellule souche hématopoïétique et permet de reconstituer un système hématopoïétique fonctionnel, associé à une amélioration clinique évidente. Par exemple, la reconstitution d’un système hématopoïétique normal et la disparition des symptômes, survenant à la suite d’un processus de chromothripsis ayant éliminé l’allèle muté, ont été observées chez un patient atteint du syndrome de WHIM causé par une mutation germinale du gène CXCR4 [41]. De manière similaire, un SGR correspondant à une réversion (back-mutation), acquise dans les cellules souches hématopoïétiques d’un patient ayant des anomalies hématologiques sévères causées par une mutation germinale homozygote de MYSM1, a permis une normalisation progressive de tous les paramètres sanguins [15, 51]. De façon remarquable, plus de sept ans après la détection de ce SGR, le patient ne présente plus aucun défaut hématopoïétique. Des exemples similaires ont été rapportés chez des individus porteurs d’un déficit en facteurs GATA2 ou RPS19 chez lesquels un SGR a permis la reconstitution d’un système hématopoïétique normal et une disparition des symptômes hématologiques [16, 38].
Ces rares exemples de SGR ayant permis la restauration d’un système hématopoïétique stable et fonctionnel peuvent, par conséquent, être qualifiés d’évènements de thérapie génique naturelle.
Potentiels effets négatifs du sauvetage génétique somatique
La présence d’un SGR chez un patient atteint d’une maladie hématopoïétique mendélienne peut parfois avoir des conséquences négatives. En effet, si le SGR est associé à une amélioration ou à une modification partielle des symptômes, celui-ci peut compliquer la compréhension de la maladie et, par conséquent, retarder le diagnostic clinique et la prise en charge adéquate du patient. Par ailleurs, les SGR associés à l’élimination de la mutation (monosomie, délétion, réversion, conversion génique) (Figure 2 et Figure 3) peuvent compromettre le diagnostic moléculaire. En effet, si le degré de mosaïcisme du SGR est important, le séquençage de l’ADN extrait des cellules sanguines peut ne pas détecter la mutation germinale puisque cette dernière aura été éliminée par le SGR. C’est pourquoi, lorsqu’on recherche la cause génétique d’une maladie hématopoïétique héréditaire, il est préférable d’utiliser de l’ADN extrait de tissus non hématopoïétiques, ces derniers ne sont pas porteurs d’un éventuel SGR [12, 18].
En outre, si le SGR est acquis dans une cellule très différenciée, cela peut provoquer des effets néfastes. C’est le cas, par exemple, des déficits immunitaires héréditaires associés à un défaut de production des lymphocytes T. Si le SGR apparaît dans un précurseur de lymphocytes T et provoque une expansion clonale, la population lymphocytaire qui en découlera n’aura pas un répertoire antigénique suffisamment diversifié et ne sera pas correctement régulée. Cela peut conduire à des réactions auto-immunes provoquant une inflammation sévère de la peau, de l’intestin ou du foie. Ce phénomène a été observé chez des patients atteints de déficits immunitaires combinés sévères [52]. Un autre exemple de SGR ayant un effet néfaste est la monosomie 7 observée dans des clones de cellules sanguines issues de patients atteints de syndromes liés aux gènes SAMD9/SAMD9L. En effet, dans ce contexte, la monosomie élimine la mutation germinale (dans SAMD9 ou SAMD9L) responsable du syndrome mais également bien d’autres gènes portés par le chromosome 7. Or, la monosomie 7 prédispose au développement de syndromes myélodysplasiques et à l’acquisition de mutations oncogéniques responsables de cancer. Ainsi la monosomie 7 dans les syndromes liés aux gènes SAMD9/SAMD9L représente un SGR bénéfique au niveau cellulaire mais possiblement associé à une évolution défavorable pour le patient sur le long terme [17, 34, 53].
Conclusions et perspectives
L’identification de SGR dans un nombre croissant de maladies hématopoïétiques à hérédité mendélienne permet de mieux comprendre la fonction des gènes impliqués et des mécanismes altérés dans ces maladies. Comme évoqué précédemment, la détection de SGR dans ces maladies impose une surveillance accrue des patients afin de s’assurer que ces SGR ne provoquent pas d’effets adverses à long terme. À l’inverse, l’observation dans certaines maladies (syndrome de WHIM, défaut en GATA2 ou MYSM1), d’un effet bénéfique provoqué par un SGR apparu dans une cellule souche hématopoïétique ouvre des perspectives thérapeutiques. En effet, dans ce type de maladie, l’avantage sélectif conféré par le SGR suggère que des stratégies d’édition de gène à visée thérapeutique (thérapie génique), même sur un nombre limité de cellules souches hématopoïétiques, pourraient être efficaces et permettre de reconstituer un système hématopoïétique stable et fonctionnel.
L’identification de SGR peut aussi inspirer de nouvelles stratégies thérapeutiques. Par exemple, il a été démontré que des mutations somatiques ponctuelles affectant l’affinité d’eIF6 pour la sous-unité pré-60S (Figure 4) représentaient des SGR indirects dans le syndrome de Shwachman-Diamond. Ce type de SGR a par ailleurs été associé à des améliorations hématologiques chez certains patients et à un sauvetage phénotypique dans un modèle de drosophile déficient en SBDS [46]. Ces observations ont conduit des laboratoires à développer des molécules qui miment l’effet de ces SGR en perturbant l’interaction d’eIF6 avec la sous-unité pré-60S [54]. Théoriquement, ces molécules pourraient présenter des vertus thérapeutiques dans le syndrome de Shwachman-Diamond, mais leurs effets spécifiques dans le contexte de cette maladie n’ont pas encore été rapportés.
Cette synthèse s’est focalisée sur le phénomène de SGR dans le contexte de maladies à hérédité mendélienne affectant le système hématopoïétique, dans lesquelles ces mécanismes ont été le plus fréquemment décrits. Cependant, quelques cas de SGR ont été rapportés dans des maladies héréditaires affectant d’autres tissus et organes. Ainsi, les SGR qui surviennent dans le cadre de certaines maladies dermatologiques génèrent de petites zones de peau saine correspondant aux zones d’expansion clonale des cellules somatiquement modifiées [55, 56]. D’autres cas de SGR dans des maladies non hématopoïétiques ont été décrits, incluant la dystrophie musculaire de Duchenne [57-60] et la maladie hépatique associée à un déficit en alpha-1 antitrypsine [61].
Enfin, il a été récemment rapporté que l’expression clinique de certaines maladies à hérédité mendélienne pouvait être modulée par des modifications des marques épigénétiques [62]. Il est par conséquent envisageable que des changements épigénétiques, tels que la méthylation de l’ADN ou les modifications post-traductionnelles des histones, puissent compenser l’impact délétère d’une mutation germinale et ainsi représenter des phénomènes de sauvetage épigénétique somatique (somatic epigenetic rescue).
En conclusion, le phénomène de sauvetage génétique somatique représente probablement un processus universel des maladies à hérédité mendélienne. Le développement de techniques de plus en plus sensibles et fiables de séquençage de l’ADN permettra vraisemblablement d’identifier des SGR dans un nombre croissant de maladies affectant de nombreux tissus et organes différents. À l’avenir, la surveillance des malades par un suivi des SGR ainsi qu’une meilleure connaissance des SGR et de leurs impacts devraient permettre de mieux comprendre l’évolution clinique des patients, d’affiner le diagnostic génétique et de développer des stratégies thérapeutiques originales.
Remerciements
Ce travail a bénéficié du soutien de la ligue nationale contre le Cancer (Laboratoire Labellisé Ligue 2023) ainsi que d’un financement ANR PRCI (Muta Z8). Sophie de Tocqueville a bénéficié d’une bourse de thèse du Ministère de l’enseignement supérieur, de la recherche et de l’industrie (MESRI) ainsi qu’une bourse de 4e année de thèse de La Ligue contre le Cancer. Patrick Revy est un chercheur du Centre National de la Recherche Scientifique (CNRS).
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Vijg J, Dong X. Pathogenic Mechanisms of somatic mutation and genome mosaicism in aging. Cell 2020; 182 : 12–23. [Google Scholar]
- Yoshida M, Tanase-Nakao K, Shima H, et al. Prevalence of germline GATA2 and SAMD9/9L variants in paediatric haematological disorders with monosomy 7. Br J Haematol 2020; 191 : 835–43. [Google Scholar]
- Moore L, Cagan A, Coorens THH, et al. The mutational landscape of human somatic and germline cells. Nature 2021; 597 : 381–6. [Google Scholar]
- Machado HE, Obro NF, Williams N, et al. Convergent somatic evolution commences in utero in a germline ribosomopathy. Nat Commun 2023; 14 : 5092. [Google Scholar]
- Abascal F, Harvey LMR, Mitchell E, et al. Somatic mutation landscapes at single-molecule resolution. Nature 2021; 593 : 405–10. [Google Scholar]
- Blokzijl F, de Ligt J, Jager M, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016; 538 : 260–4. [Google Scholar]
- Jacobs KB, Yeager M, Zhou W, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet 2012; 44 : 651–8. [Google Scholar]
- Laurie CC, Laurie CA, Rice K, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet 2012; 44 : 642–50. [Google Scholar]
- Lee-Six H, Obro NF, Shepherd MS, et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 2018; 561 : 473–8. [Google Scholar]
- Osorio FG, Rosendahl Huber A, Oka R, et al. Somatic Mutations reveal lineage relationships and age-related mutagenesis in human hematopoiesis. Cell Rep 2018; 25 : 2308–16 e4. [Google Scholar]
- Forterre P. Darwin’s goldmine is still open: variation and selection run the world. Front Cell Infect Microbiol 2012; 2 : 106. [Google Scholar]
- Revy P, Kannengiesser C, Fischer A. Somatic genetic rescue in Mendelian haematopoietic diseases. Nat Rev Genet 2019; 20 : 582–98. [Google Scholar]
- Mitchell E, Spencer Chapman M, Williams N, et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature 2022; 606 : 343–50. [Google Scholar]
- Gutierrez-Rodrigues F, Groarke EM, Thongon N, et al. Clonal landscape and clinical outcomes of telomere biology disorders: somatic rescuing and cancer mutations. Blood 2024 ; 144 : 2402–16. [Google Scholar]
- Le Guen T, Touzot F, Andre-Schmutz I, et al. An in vivo genetic reversion highlights the crucial role of Myb-Like, SWIRM, and MPN domains 1 (MYSM1) in human hematopoiesis and lymphocyte differentiation. J Allergy Clin Immunol 2015; 136 : 1619–26. [Google Scholar]
- Catto LFB, Borges G, Pinto AL, et al. Somatic genetic rescue in hematopoietic cells in GATA2 deficiency. Blood 2020; 136 : 1002–5. [Google Scholar]
- Sahoo SS, Pastor VB, Goodings C, et al. Clinical evolution, genetic landscape and trajectories of clonal hematopoiesis in SAMD9/SAMD9L syndromes. Nat Med 2021; 27 : 1806–17. [Google Scholar]
- Bluteau O, Sebert M, Leblanc T, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood 2018; 131 : 717–32. [Google Scholar]
- Buonocore F, Kuhnen P, Suntharalingham JP, et al. Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans. J Clin Invest 2017; 127 : 1700–13. [Google Scholar]
- Shima H, Koehler K, Nomura Y, et al. Two patients with MIRAGE syndrome lacking haematological features: role of somatic second-site reversion SAMD9 mutations. J Med Genet 2018; 55 : 81–5. [Google Scholar]
- Tesi B, Davidsson J, Voss M, et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood 2017; 129 : 2266–79. [Google Scholar]
- Revy P, Kannengiesser C, Bertuch AA. Genetics of human telomere biology disorders. Nature Rev Genet 2023; 24 : 86–108. [Google Scholar]
- Hoareau-Aveilla C, Henry Y, Leblanc T. Dyskératose congénitale, une maladie causée par une maintenance défectueuse des télomères. Med Sci (Paris) 2008; 24 : 390–8. [Google Scholar]
- Hennion N, Desseyn JL, Gottrand F, et al. Fibrose pulmonaire idiopathique. Med Sci (Paris) 2022; 38 : 579–84. [Google Scholar]
- Gutierrez-Rodrigues F, Donaires FS, Pinto A, et al. Pathogenic TERT promoter variants in telomere diseases. Genet Med 2019; 21 : 1594–602. [Google Scholar]
- Maryoung L, Yue Y, Young A, et al. Somatic mutations in telomerase promoter counterbalance germline loss-of-function mutations. J Clin Invest 2017; 127 : 982–6. [Google Scholar]
- Benyelles M, O’Donohue MF, Kermasson L, et al. NHP2 deficiency impairs rRNA biogenesis and causes pulmonary fibrosis and Hoyeraal-Hreidarsson syndrome. Hum Mol Genet 2020; 29 : 907–22. [Google Scholar]
- Bertrand A, Ba I, Kermasson L, et al. Characterization of novel mutations in the TEL-patch domain of the telomeric factor TPP1 associated with telomere biology disorders. Hum Mol Genet 2024; 20 : 612–23. [Google Scholar]
- Ellis NA, Ciocci S, German J. Back mutation can produce phenotype reversion in Bloom syndrome somatic cells. Hum Genet 2001; 108 : 167–73. [Google Scholar]
- Alder JK, Stanley SE, Wagner CL, et al. Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest 2015; 147 : 1361–8. [Google Scholar]
- Wada T, Schurman SH, Otsu M, et al. Somatic mosaicism in Wiskott-Aldrich syndrome suggests in vivo reversion by a DNA slippage mechanism. Proc Nat Acad Sci USA 2001; 98 : 8697–702. [Google Scholar]
- Cunniff C, Bassetti JA, Ellis NA. Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol Syndromol 2017; 8 : 4–23. [Google Scholar]
- Venugopal P, Arts P, Fox LC, et al. Unraveling facets of MECOM-associated syndrome: somatic genetic rescue, clonal hematopoiesis, and phenotype expansion. Blood Adv 2024; 8 : 3437–43. [Google Scholar]
- Wong JC, Bryant V, Lamprecht T, et al. Germline SAMD9 and SAMD9L mutations are associated with extensive genetic evolution and diverse hematologic outcomes. JCI Insight 2018; 3 : e121086. [Google Scholar]
- Sharma R, Sahoo SS, Honda M, et al. Gain-of-function mutations in RPA1 cause a syndrome with short telomeres and somatic genetic rescue. Blood 2022; 139 : 1039–51. [Google Scholar]
- Jongmans MCJ, Diets IJ, Quarello P, et al. Somatic reversion events point towards RPL4 as a novel disease gene in a condition resembling Diamond-Blackfan Anemia. Haematologica 2018; 103 : e607–9. [Google Scholar]
- Jongmans MC, Verwiel ET, Heijdra Y, et al. Revertant somatic mosaicism by mitotic recombination in dyskeratosis congenita. Am J Hum Genet 2012; 90 : 426–33. [Google Scholar]
- Garelli E, Quarello P, Giorgio E, et al. Spontaneous remission in a Diamond-Blackfan anaemia patient due to a revertant uniparental disomy ablating a de novo RPS19 mutation. Brit J Haematol 2019; 185 : 994–8. [Google Scholar]
- Sahoo SS, Erlacher M, Wlodarski MW. Genetic and clinical spectrum of SAMD9 and SAMD9L syndromes: from variant interpretation to patient management. Blood 2025; 145 : 475–85. [Google Scholar]
- Pellestor F, Gatinois V, Puechberty J, et al. La chromothripsis, une nouvelle forme inattendue de complexité pour les réarrangements chromosomiques. Med Sci (Paris) 2014; 30 : 266–73. [Google Scholar]
- McDermott DH, Gao JL, Liu Q, et al. Chromothriptic cure of WHIM syndrome. Cell 2015; 160 : 686–99. [Google Scholar]
- Warren AJ. Molecular basis of the human ribosomopathy Shwachman-Diamond syndrome. Adv Biol Regul 2017; S2212-4926 : 30153–7. [Google Scholar]
- Pressato B, Valli R, Marletta C, et al. Deletion of chromosome 20 in bone marrow of patients with Shwachman-Diamond syndrome, loss of the EIF6 gene and benign prognosis. Brit J Haematol 2012; 157 : 503–5. [Google Scholar]
- Valli R, Minelli A, Galbiati M, et al. Shwachman-Diamond syndrome with clonal interstitial deletion of the long arm of chromosome 20 in bone marrow: haematological features, prognosis and genomic instability. Brit J Haematol 2019; 184 : 974–81. [Google Scholar]
- Valli R, Pressato B, Marletta C, et al. Different loss of material in recurrent chromosome 20 interstitial deletions in Shwachman-Diamond syndrome and in myeloid neoplasms. Mol Cytogenet 2013; 6 : 56. [Google Scholar]
- Tan S, Kermasson L, Hilcenko C, et al. Somatic genetic rescue of a germline ribosome assembly defect. Nat Commun 2021; 12 : 5044. [Google Scholar]
- Kennedy AL, Myers KC, Bowman J, et al. Distinct genetic pathways define pre-malignant versus compensatory clonal hematopoiesis in Shwachman-Diamond syndrome. Nat Commun 2021; 12 : 1334. [Google Scholar]
- Schratz KE, Gaysinskaya V, Cosner ZL, et al. Somatic reversion impacts evolution of myelodysplastic syndromes and acute myeloid leukemia in the short telomere disorders. J Clin Invest 2021 ; 131 : e147598. [Google Scholar]
- Kochman R, Ba I, Yates M, et al. Heterozygous RPA2 variant as a novel genetic cause of telomere biology disorders. Genes Dev 2024; 38 : 755–71. [Google Scholar]
- Sande CM, Chen S, Mitchell DV, et al. ATM-dependent DNA damage response constrains cell growth and drives clonal hematopoiesis in telomere biology disorders. J Clin Invest 2025; 135. [Google Scholar]
- de Tocqueville S, Martin E, Riller Q, et al. Long-term assessment of haematological recovery following somatic genetic rescue in a MYSM1-deficient patient: Implications for in vivo gene therapy. Brit J Haematol 2024; 205 : 2349–54. [Google Scholar]
- Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol 2016; 16 : 234–46. [Google Scholar]
- Davidsson J, Puschmann A, Tedgard U, et al. SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 2018; 32 : 1106–15. [Google Scholar]
- Pesce E, Miluzio A, Turcano L, et al. discovery and preliminary characterization of translational modulators that impair the binding of eIF6 to 60S ribosomal subunits. Cells 2020; 9 : 172. [Google Scholar]
- Shipman AR, Liu L, Lai-Cheong JE, et al. Somatic forward (nonrevertant) mosaicism in recessive dystrophic epidermolysis bullosa. JAMA Dermatol 2014; 150 : 1025–7. [Google Scholar]
- Lim YH, Fisher JM, Choate KA. Revertant mosaicism in genodermatoses. Cell Mol Life Sci 2017; 74 : 2229–38. [Google Scholar]
- Burrow KL, Coovert DD, Klein CJ, et al. Dystrophin expression and somatic reversion in prednisone-treated and untreated Duchenne dystrophy. CIDD Study Group. Neurology 1991; 41 : 661–6. [Google Scholar]
- Fanin M, Danieli GA, Cadaldini M, et al. Dystrophin-positive fibers in Duchenne dystrophy: origin and correlation to clinical course. Muscle Nerve 1995; 18 : 1115–20. [Google Scholar]
- Klein CJ, Coovert DD, Bulman DE, et al. Somatic reversion/suppression in Duchenne muscular dystrophy (DMD): evidence supporting a frame-restoring mechanism in rare dystrophin-positive fibers. Am J Hum Genet 1992; 50 : 950–9. [Google Scholar]
- Punetha J, Mansoor S, Bertorini TE, et al. Somatic mosaicism due to a reversion variant causing hemi-atrophy: a novel variant of dystrophinopathy. Eur J Hum Genet 2016; 24 : 1511–4. [Google Scholar]
- Brzozowska N, Wu LYD, Khodzhaeva V, et al. Selection for somatic escape variants in SERPINA1 in the liver of patients with alpha-1 antitrypsin deficiency. Nat Genet 2025; 57 : 875–83. [Google Scholar]
- Stewart O, Gruber C, Randolph HE, et al. Monoallelic expression can govern penetrance of inborn errors of immunity. Nature 2025; 637 : 1186–97. [Google Scholar]
- Gilgenkrantz H. Trans… Med Sci (Paris) 2024 ; 40 : 555. [Google Scholar]
Du grec : chromos pour chromosome et thripsis pour briser en éclats
Le syndrome WHIM est une maladie immunologique héréditaire à transmission autosomique dominante. Son nom correspond à l’acronyme anglophone dérivé de « warts (verrues), hypogammaglobulinemia, immunodeficiency, myelokathexis (leucopénie et neutropénie dues à une rétention de ces cellules dans la moelle) ». C’est une maladie extrêmement rare (ndlr).
Liste des figures
|
Figure 1 Principe du sauvetage génétique somatique. Dans le contexte d’une maladie à hérédité mendélienne (avec cellules porteuses d’une mutation germinale délétère), une mutation somatique peut avoir plusieurs effets : A. elle peut être délétère et conduire à la disparition de la cellule par apoptose ou immunosurveillance ; B. elle peut être neutre, et les cellules porteuses de la mutation somatique se comporteront comme les cellules non modifiées ; C. elle peut contrer l’effet délétère de la mutation germinale et conférer un avantage sélectif aux cellules somatiquement modifiées et provoquer une expansion clonale. Ce dernier cas correspond au mécanisme de sauvetage génétique somatique. Les cas B. et C. génèrent du mosaïcisme cellulaire. Les cellules portant une mutation somatique sont représentées avec un noyau orange. La membrane lobée représente les cellules dont la fonction est altérée par la mutation germinale |
| Dans le texte | |
|
Figure 2 Les différentes possibilités de mutations ponctuelles associées aux SGR. |
| Dans le texte | |
|
Figure 3 Exemples d’événements génétiques somatiques associés à des SGR directs. Dans le contexte d’une maladie à hérédité mendélienne causée par une mutation germinale hétérozygote avec effet dominant négatif (A.), un SGR direct peut survenir par : B. mutation ponctuelle qui remplace la mutation germinale par la séquence sauvage (réversion) ; C. mutation ponctuelle non-sens qui élimine l’expression du gène portant la mutation germinale ; D. conversion génique qui copie une partie du chromosome sauvage à la place du gène portant la mutation germinale (disomie uniparentale); E. perte du chromosome (monosomie) contenant le gène portant la mutation germinale; F. délétion interstitielle éliminant une portion du chromosome incluant le gène portant la mutation germinale. |
| Dans le texte | |
|
Figure 4 Exemple de SGR indirect dans le contexte du syndrome de Shwachman-Diamond. A. SBDS est une protéine qui retire le facteur eIF6 de la sous-unité ribosomale pré-60S afin de permettre son interaction avec la sous-unité pré-40S pour former un ribosome fonctionnel. B. Des mutations germinales délétères de SBDS affectent le mécanisme d’éviction d’eIF6, la formation de ribosome et causent le syndrome de Shwachman-Diamond. C. une mutation somatique dans le gène EIF6 (ici est représentée une délétion interstitielle) qui affecte l’expression ou la fonction anti-associative d’eIF6 représente un SGR en restaurant la formation de ribosomes fonctionnels. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.