")
")
| Issue |
Med Sci (Paris)
Volume 40, Number 12, Décembre 2024
Épigénétique : développement et destin cellulaire
|
|
|---|---|---|
| Page(s) | 955 - 962 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2024182 | |
| Published online | 20 December 2024 | |
L’ingénierie ciblée de l’épigénome
Targeted epigenome engineering
1
Institut Curie, Paris Sciences et Lettres, Sorbonne Université, Paris, France
2
Inserm U934/CNRS UMR 3215, Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
La différenciation et l’homéostasie cellulaires reposent sur des mécanismes élaborés de contrôle de l’expression des gènes, qui permettent aux différents lignages cellulaires d’un organisme d’établir puis de « mémoriser » différents états épigénétiques. Les processus qui contrôlent l’expression des gènes sont centrés sur la chromatine, un complexe composé d’ADN, de protéines histones et d’ARN, dont la structure est finement régulée. Les outils d’ingénierie de l’épigénome permettent d’interférer avec ces processus et de les étudier, révélant la logique des mécanismes de mémoire épigénétique. Cet article présente les principales classes d’outils de modification ciblée de l’épigénome et illustre comment ils peuvent être utilisés afin de mieux comprendre et modifier l’épigénome des cellules, ouvrant la voie à des perspectives thérapeutiques révolutionnaires.
Abstract
Cellular differentiation and homeostasis rely on complex mechanisms to control gene expression, enabling the different cell lineages of an organism to establish and then “memorize” different epigenetic states. The processes that control gene expression are centered on chromatin, a complex of DNA, histone proteins and RNA, whose structure is finely regulated. Targeted epigenomic engineering tools make it possible to interfere with and study these processes, revealing the logic of epigenetic memory mechanisms. This article reviews the main classes of targeted epigenome modification tools and illustrates how they can be used to better understand and modify the epigenome of cells, paving the way for potentially revolutionary therapeutic prospects.
© 2024 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (© Biorender.com).
Les organismes pluricellulaires sont constitués de dizaines à plusieurs centaines de types cellulaires différents, assemblés au sein de tissus et d’organes qui assurent les fonctions vitales de l’organisme. L’étude des processus qui sous-tendent la génération de cette multitude de cellules spécialisées est un sujet fondamental en biologie, qui a des implications médicales importantes. Une compréhension approfondie des mécanismes de différenciation cellulaire permet, par exemple, d’envisager un contrôle de la production de types cellulaires particuliers à des fins thérapeutiques [1]. Les événements moléculaires qui guident la génération des différents types cellulaires ne peuvent être directement déduits de la séquence d’ADN [2]. En effet, en dehors de quelques exceptions, toutes les cellules d’un organisme contiennent le même ADN. Une partie importante de ce qui les distingue les unes des autres tient aux batteries de gènes qu’elles expriment ou n’expriment pas, si bien qu’à un génome donné peuvent correspondre une multitude d’« épigénomes », chacun caractérisé par une combinaison spécifique de gènes exprimés. Une part importante de l’activité de recherche contemporaine s’attache donc à élucider les mécanismes qui permettent la mise en place puis le maintien de ces épigénomes au sein des différents lignages cellulaires.
Les études menées au cours des dernières décennies nous ont appris que la transcription, première étape de l’expression des gènes, est un processus complexe faisant intervenir plusieurs niveaux de régulation. L’ADN des cellules eucaryotes est étroitement associé à des protéines histones [3], dont la présence constitue un masque qui restreint la disponibilité ou l’« accessibilité » de l’ADN vis-à-vis des protéines régulatrices. La fibre chromatinienne ainsi formée est le siège d’un premier niveau de régulation : des enzymes, qui ajoutent des groupements chimiques sur la chromatine ou qui déplacent ou éjectent les histones, modulent l’accessibilité du matériel génétique, le prédisposant soit à devenir actif, soit, au contraire, à rester inactif [4]. Un second niveau de régulation peut alors se déployer : un gène de la fraction accessible de la chromatine devient « exprimé » si des protéines « activatrices » du noyau se lient à certaines portions dites « régulatrices » de son ADN. À l’inverse, des protéines « répressives » peuvent inactiver l’expression d’un gène. Ces protéines qui régulent l’expression génique sont les facteurs de transcription [5]. Il existe un lien de causalité réciproque entre ces deux niveaux de régulation. En effet, la liaison d’un facteur de transcription sur une séquence du génome peut à son tour défavoriser la liaison des histones à l’ADN et donc accroître l’accessibilité de la chromatine. En outre, de nombreux facteurs de transcription agissent en recrutant des enzymes de modification de la chromatine, dont l’action peut également modifier l’accessibilité locale de la chromatine [6,7]. D’autres entités moléculaires, comme des ARN non-codants, participent à la régulation de l’expression génique selon diverses modalités, par exemple, en assurant le recrutement de facteurs de transcription et/ou de régulateurs chromatiniens, en interférant avec la transcription, etc. Les liens entre régulation de la structure chromatinienne, liaison de facteurs de transcription et action des ARN non-codants, ainsi que la façon dont ces interactions se déploient dans l’espace tridimensionnel de la fibre chromatinienne, font actuellement l’objet de nombreuses études.
Les outils d’ingénierie de l’épigénome élargissent considérablement les possibilités d’analyse de ces phénomènes. En effet, en nous donnant les moyens d’interférer avec les différentes entités impliquées, ces outils nous permettent d’interroger le fonctionnement et la dynamique des processus moléculaires. Les « réponses » qui résultent de ces expérimentations nous permettent d’affiner notre compréhension de la façon dont l’expression des gènes est régulée. En fonction de leur niveau d’action, les approches expérimentales développées en laboratoire peuvent schématiquement être subdivisées en deux catégories. Un premier type d’approche d’interférence « globale » consiste à supprimer (et/ou plus rarement à augmenter) l’activité d’une entité moléculaire, par exemple une enzyme de modification de la chromatine. Les effets produits par ces interférences sont mesurés à l’aide d’analyses reposant sur le séquençage à haut débit. Elles permettent d’évaluer l’ensemble des changements d’expression (par séquençage de l’ARN, RNA-seq), de composition (en déterminant les interactions entre ADN et protéines, ChIP-seq ou CUT&RUN-seq) ou d’accessibilité de la chromatine (ATAC-seq), de topologie tridimensionnelle (Hi-C), etc. [8]. C’est en corrélant la localisation initiale de l’entité moléculaire étudiée et l’effet de sa suppression (ou de son augmentation, le cas échéant) que l’on peut inférer sa fonction dans la régulation de l’expression génique. Or, la possibilité d’établir un lien de causalité clair est souvent compliquée par la survenue d’effets secondaires, par exemple, dans le cas où les produits de gènes dont l’expression est altérée modifient à leur tour l’expression d’autres gènes.
Un second type d’approche, dite « locale », vise à restreindre l’interférence à une région précise du génome, ce qui permet, en principe, de limiter les effets secondaires. Les approches locales nécessitent le développement d’outils permettant de diriger une entité moléculaire d’intérêt, par exemple le domaine catalytique d’une enzyme de modification de la chromatine, vers une séquence d’ADN spécifique du génome. C’est pourquoi les approches locales ont initialement ciblé des gènes artificiels (ou transgènes) constitués de séquences d’ADN constantes, susceptibles d’être liées par des modules protéiques définis. En fusionnant des domaines protéiques d’intérêt à ces modules de liaison à l’ADN, il est possible de les adresser spécifiquement à l’ADN du transgène et d’étudier leur influence sur l’expression et/ou sur les propriétés de la chromatine [9,10]. Les outils d’ingénierie de l’épigénome permettent d’étendre les approches d’analyse locale à des séquences d’ADN naturellement présentes dans le génome. Outre leur intérêt en recherche fondamentale, ces outils ouvrent de nouvelles perspectives pour la reprogrammation de l’épigénome cellulaire à des fins thérapeutiques.
Après un bref historique des méthodes d’ingénierie de l’épigénome, nous aborderons les différentes classes d’outils qui ont été développées. En nous appuyant sur des exemples de la littérature scientifique, nous illustrerons l’apport de ces outils à l’analyse des phénomènes épigénétiques et les applications susceptibles d’en découler.
Une brève histoire des outils d’ingénierie épigénétique
Le développement des techniques de modifications ciblées de l’épigénome est étroitement lié à celui des approches de manipulation génétique. Ces deux types d’approches reposent sur la capacité à diriger tout ou partie d’une protéine vers une séquence d’ADN spécifique. Cette opération nécessite la mise au point d’un système moléculaire de liaison à l’ADN qui soit « programmable », c’est-à-dire capable de se fixer sur une séquence d’ADN définie à l’avance. Dès lors qu’un module programmable de liaison à l’ADN existe, il est possible d’y accoler un domaine protéique permettant de modifier l’ADN cible ou son environnement (Figure 1). Si l’objectif est la manipulation génétique, on utilise des domaines enzymatiques qui catalysent la coupure de l’ADN ou la modification directe de la séquence nucléotidique. S’il s’agit de modifier l’expression et/ou la composition de la chromatine, on a recours à des domaines de modulation de la transcription et/ou de la chromatine [11,12].
 |
Figure 1 Les principales familles d’outils d’ingénierie épigénétique. A. Zinc Fingers (ZF) – Les domaines ZF reconnaissent une séquence d’ADN spécifique et permettent d’y cibler un domaine effecteur. B. TALE – Le domaine effecteur est lié au domaine de reconnaissance et de liaison à l’ADN composé d’arrangements variables de modules liant chacun un des quatre nucléotides de l’ADN. C. Système CRISPR-dCas9 – L’ARN guide s’apparie à la séquence cible et permet l’amarrage de la protéine dCas9 fusionnée au domaine effecteur. |
Les premiers efforts de développement de domaines programmables de liaison à l’ADN
La mise au point des premiers domaines programmables de liaison à l’ADN date des années 1990 et est le fruit d’un long travail d’ingénierie, qui utilise des domaines protéiques de liaison à l’ADN de type « doigt de zinc » (zinc fingers, ZF), naturellement présents dans de nombreux facteurs de transcription [13]. Il existe une multitude de domaines ZF ayant différentes spécificités de liaison à l’ADN. Un domaine ZF reconnaît trois paires de bases de l’ADN, et la séquence reconnue varie d’un domaine à l’autre. L’assemblage de plusieurs de ces domaines permet de créer une protéine chimérique capable de lier une séquence d’ADN spécifique du génome. De nombreuses études ont démontré l’utilité de domaines ZF fusionnés à une enzyme de coupure de l’ADN pour des expériences de modification ciblée du génome [14]. Les domaines ZF ont également donné naissance à des expériences pionnières d’ingénierie de l’épigénome, que ce soit pour le contrôle de l’expression génique [15] ou pour des modifications ciblées de la chromatine [16,17]. Ainsi, l’approche fondée sur les domaines ZF a posé les bases conceptuelles et expérimentales de l’ingénierie de l’épigénome. Cette méthode de ciblage génétique est laborieuse à mettre en œuvre et souffre de plusieurs limitations, notamment de la difficulté de prédire avec précision leur spécificité de liaison à l’ADN, ce qui a grandement freiné la diffusion de cet outil au sein des laboratoires. Ces limitations ont été en partie contournées avec la découverte des protéines de type TALE (transcription activator-like effector), qui sont des facteurs de transcription présents chez les phytobactéries pathogènes du genre Xanthomonas [18,19]. Le domaine de liaison à l’ADN des protéines TALE est composé d’arrangements variables de modules liant chacun un des quatre nucléotides de l’ADN. Un domaine capable de lier une nouvelle séquence peut ainsi être créé en assemblant une série de modules selon l’ordre prescrit par l’ADN à cibler. Les approches d’édition du génome ou de l’épigénome fondées sur les domaines ZF ont rapidement été transposées au système TALE [20,21].
L’outil CRISPR-Cas9 et l’essor des approches d’édition du génome et de l’épigénome
Peu après le développement des approches fondées sur les TALE, un nouvel outil est apparu, appelé CRISPR-Cas9, qui multiplie les possibilités d’édition du génome et de l’épigénome. Découvert chez la bactérie Streptococcus pyogenes, CRISPR-Cas9 est un système de défense élaboré par les bactéries pour lutter contre leurs virus, les bactériophages [22]. Ce système repose sur l’interaction entre la nucléase Cas9 et un ARN « guide » de vingt nucléotides, lequel est complémentaire d’une séquence d’ADN présente dans le génome du bactériophage. La liaison du complexe formé par Cas9 et l’ARN guide sur la séquence cible entraîne la coupure de l’ADN cible, et donc l’inactivation du génome du bactériophage. Le caractère révolutionnaire de cette nucléase programmable est lié à la facilité avec laquelle elle peut être programmée puisque sa spécificité de séquence repose sur l’ARN guide, entité moléculaire bien plus facile à générer que des assemblages multimériques de domaines ZF ou TALE.
Le système CRISPR-Cas9 a rapidement été adapté pour élaborer des outils de modification ciblée de l’épigénome. Dans ces approches, l’activité nucléase de la protéine Cas9 est abolie par une mutation. La protéine catalytiquement inerte, appelée dCas9, pour « dead Cas9 », reste capable de lier une séquence d’ADN en présence de l’ARN guide complémentaire. Reprenant la logique modulaire employée avec les domaines ZF et TALE, des outils d’édition de l’épigénome ont été créés en fusionnant des domaines de modulation de la transcription ou de la chromatine avec la protéine dCas9. Comme il est relativement facile d’exprimer plusieurs ARN guides simultanément, les approches d’édition de l’épigénome fondées sur CRISPR-Cas9 permettent en outre de cibler simultanément plusieurs séquences d’ADN.
De nombreux systèmes CRISPR présents dans d’autres espèces de bactéries ont été identifiés [23]. Les nouveaux outils qui résultent de ces recherches multiplient les possibilités de modulations orthogonales de l’épigénome, par exemple pour réprimer l’expression d’un premier gène tout en stimulant l’expression d’un second.
Les outils d’édition de l’épigénome
L’arrivée de l’outil CRISPR a permis de nombreuses avancées dans le domaine de l’ingénierie épigénétique. La protéine dCas9 est devenue la plateforme de choix pour recruter un domaine protéique d’intérêt au niveau d’une séquence nucléotidique. Les méthodes d’ingénierie de l’épigénome, auparavant limitées à des expérimentations conceptuelles avec les domaines ZF et les TALE, sont entrées dans la boîte à outils du laboratoire grâce au système CRISPR. Une multitude de domaines enzymatiques ou structuraux peuvent ainsi être adressés précisément sur l’ADN, décuplant les possibilités d’expérimentation locale. Nous nous intéresserons plus particulièrement à deux familles d’outils qui permettent soit de moduler la transcription, soit de modifier sélectivement la composition de la chromatine.
Régulation locale de la transcription
La fusion de la protéine dCas9 avec des domaines protéiques régulant l’expression de gènes a permis de générer des outils très efficaces d’inhibition ou d’activation locale de la transcription. Ils sont appelés respectivement « CRISPR interference » (CRISPRi, [24]) et « CRISPR activation » (CRISPRa, [25]). Le système CRISPRi, majoritairement employé, utilise une fusion entre dCas9 et le domaine répresseur appelé KRAB (Krüppel-associated box) (Figure 2A). Ce type de domaine est présent dans une vaste famille de facteurs de transcription impliqués dans la répression transcriptionnelle, en particulier celles des éléments transposables. Le domaine KRAB interagit avec la protéine KAP1 (KRAB-associated protein-1) qui, elle-même, interagit avec plusieurs régulateurs chromatiniens (notamment CBX5/HP1a [heterochromatin protein 1 alpha], SETDB1 [SET domain bifurcated histone lysine methyltransferase 1], NuRD [nucleosome remodeling and deacetylase]), dont l’action concertée réduit l’accessibilité de la chromatine, ce qui entraîne l’inhibition de la transcription. Les systèmes CRISPRa, inversement, utilisent des domaines d’activation de la transcription issus de facteurs de transcription activateurs (Figure 2B). Ces domaines agissent en favorisant le recrutement de la machinerie transcriptionnelle ainsi que celui de régulateurs chromatiniens (par exemple des histones acétyl-transférases ou des remodeleurs de la chromatine) qui établissent un état chromatinien accessible, et donc transcriptionnellement permissif. Parmi les différents systèmes CRIS-PRa développés, ceux qui combinent plusieurs domaines d’activation de la transcription sont ceux qui stimulent le plus fortement l’expression génique [26].
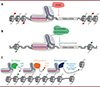 |
Figure 2 Outils d’édition de l’épigénome basés sur l’approche CRISPR/dCas9. A. La méthode « CRISPR interference » (CRISPRi) utilise une fusion de la version inactive de Cas9 (dCas9) avec le domaine KRAB, lequel induit la répression transcriptionnelle du gène cible en établissant un état chromatinien répressif. B. Le système « CRISPR activation » (CRISPRa) résulte de la fusion dCas9 avec un ou plusieurs activateurs transcriptionnels. Il permet d’activer l’expression du gène cible grâce à l’établissement d’un état chromatinien permissif et au recrutement de la machinerie transcriptionnelle. C. La fusion de différents domaines effecteurs de régulateurs épigénétiques à la protéine dCas9 permet de moduler la composition de la chromatine via l’ajout ou l’effacement de diverses modifications chimiques, ainsi que par l’ajout ou l’éjection de nucléosomes. HMT : histone méthyltransférase ; HAT : histone acétyltransférase ; HDAC : histone déacétylase ; KDM : lysine déméthylase. |
Modulation de l’activité des séquences régulatrices enhancer
Plusieurs études ont montré que l’outil CRISPRi permet, outre la modulation de l’expression de gènes individuels, la régulation de l’activité de séquences d’ADN régulatrices enhancer (amplificateur). Les séquences enhancer sont des plateformes de liaison pour des facteurs de transcription qui modulent l’expression des gènes situés à proximité. Le recrutement de la fusion dCas9-KRAB au niveau d’une séquence enhancer active, c’est-à-dire liée par des facteurs de transcription, induit une hétérochromatinisation locale, qui se traduit par une baisse d’accessibilité de la séquence enhancer [27,28]. L’hétérochromatinisation étant dans la majorité des cas restreinte à la séquence ciblée, il est possible d’inactiver les différents enhancers d’un même gène individuellement ou simultanément afin d’interroger leurs contributions respectives à la transcription du gène cible.
Les outils CRISPRa permettent, à l’inverse, de stimuler l’activité des séquences enhancer. Certaines variantes de ces outils associent notamment à la protéine dCas9 des enzymes qui catalysent des modifications d’histone enrichies aux séquences enhancer (voir par exemple [29,30]).
Modification locale de la chromatine
Afin d’analyser plus finement le rôle des modifications chromatiniennes dans la régulation transcriptionnelle, des outils qui permettent de modifier la composition de la chromatine de façon sélective sont nécessaires. De tels outils ont été développés en fusionnant la protéine dCas9 avec des domaines d’enzymes de modification de la chromatine déjà connus (Figure 2C). Ces enzymes catalysent l’écriture ou l’effacement de diverses modifications chimiques au niveau des histones (méthylation, acétylation, ou ubiquitylation) ou de l’ADN (méthylation). L’action d’un seul domaine enzymatique ne produit qu’une modification modeste de la chromatine. Plusieurs astuces moléculaires permettent d’augmenter le nombre de domaines effecteurs recrutés par la protéine dCas9. Une première approche consiste à recruter un effecteur au niveau d’une portion de l’ARN guide, en utilisant des systèmes de liaison entre séquences d’ARN et domaines protéiques particuliers. L’ajout de plusieurs de ces séquences à l’ARN guide permet d’augmenter le nombre d’effecteurs recrutés au complexe formé par l’ARN guide et la protéine dCas9. C’est l’option qui a été retenue pour le recrutement localisé d’enzymes dites « de remodelage » de la chromatine [31], qui catalysent l’ajout ou l’éjection de nucléosomes (Figure 2C). Un second système d’amplification, appelé SunTag [32], permet le recrutement de multiples copies d’un domaine effecteur au niveau de la protéine dCas9. Une équipe de l’European Molecular Biology Laboratory (EMBL) de Rome, en Italie, a récemment développé une série d’outils CRISPR d’édition de l’épigénome utilisant le système SunTag pour catalyser l’ajout de neuf modifications chromatiniennes différentes [33]. Grâce à ces outils, il a été possible d’étudier l’influence de modifications individuelles ou combinées sur l’expression génique. Les auteurs ont également associé des approches de perturbation globales, qui éliminent l’activité catalytique d’un régulateur chromatinien, avec une restauration locale de la modification d’histone correspondante. Cette approche élégante permet de tester rigoureusement les liens de cause à effet entre une modification de la chromatine et l’expression génique.
Utilisation des outils d’ingénierie de l’épigénome en recherche biomédicale
Les outils de modulation locale de la transcription et de la chromatine constituent un apport majeur dans le domaine de la recherche en épigénétique. En ouvrant la voie à des expérimentations au niveau de séquences individuelles du génome, ils élargissent considérablement les possibilités d’analyse et de manipulation des processus de régulation de l’expression génique. La possibilité de modifier l’épigénome ouvre de nouvelles perspectives pour la reprogrammation cellulaire à des fins thérapeutiques. Nous illustrerons ces aspects par quelques exemples tirés de la recherche biomédicale.
Reprogrammation trans-épigénétique
Les expériences de reprogrammation (ou transdifférenciation) cellulaire ont pour objectif la conversion d’un type cellulaire en un autre à des fins thérapeutiques. Ce type de manipulation nécessite souvent de modifier l’expression de gènes codant des facteurs de transcription qui contrôlent l’identité cellulaire. Dans les cas les plus simples, l’expression forcée d’un seul facteur de transcription peut suffire. Cependant, dans la plupart des cas, l’expression simultanée de plusieurs facteurs est requise. Les outils de modulation locale de la transcription simplifient grandement ce type de manipulation. Une équipe de l’université de Columbia, aux États-Unis, a montré qu’une approche CRISPRa peut reprogrammer de façon stable des fibroblastes embryonnaires de souris en cellules musculaires [34]. Les auteurs de cette étude ont utilisé l’outil CRISPRa pour activer transitoirement l’expression du gène Myod1 (myogenic differentiation 1), qui code un facteur de transcription de différenciation myocytaire. Cette stimulation temporaire entraîne une expression à long terme du gène et une reprogrammation cellulaire efficace. Ce phénomène de mémoire « trans-épigénétique » est lié au fait que le gène Myod1 code un facteur de transcription capable de stimuler sa propre expression. Cette boucle de rétroaction positive permet au gène de rester exprimé après une activation transitoire (Figure 3A). Dans cet exemple, le système CRISPRa permet d’obtenir un résultat équivalent à celui obtenu par surexpression de Myod1 à partir d’un système transgénique.
 |
Figure 3 Les différentes approches d’édition de l’épigénome qui permettent d’induire des changements stables d’expression génique. A. Une activation transitoire peut être maintenue de façon prolongée si le gène est soumis à un processus de mémoire trans-épigénétique (comme c’est le cas lorsque le produit du gène activé est capable de maintenir sa propre transcription) ou à un processus de mémoire cis-épigénétique, dont la logique repose sur une opposition mutuelle entre les protéines Polycomb et la transcription. B. Le recrutement transitoire d’une protéine de fusion entre dCas9, le domaine KRAB et un module de méthylation de l’ADN permet d’instaurer une répression cis-épigénétique semblable à celle existant au niveau des éléments transposables ou de certains gènes soumis à une empreinte. |
Reprogrammation cis-épigénétique
Qu’en est-il pour les gènes qui, au contraire de Myod1, ne présentent pas de possibilité d’auto-régulation ? Est-il possible d’induire des changements d’expression irréversibles de tels gènes par une action transitoire ? Des études utilisant des outils de modification ciblée de l’épigénome suggèrent des pistes prometteuses pour atteindre un tel objectif. Notre équipe s’est intéressée à des gènes dont la chromatine est maintenue dans un état transcriptionnellement silencieux par des protéines Polycomb. En utilisant des outils CRISPRa, nous avons montré qu’une activation transitoire de tels gènes permet, dans un contexte cellulaire précis, d’induire un basculement stable de leur expression, et ce, en l’absence de boucles de régulation de type trans-épigénétique [35]. Nos analyses suggèrent que le changement induit est « cis-épigénétique », c’est-à-dire que l’information épigénétique se propage localement au niveau de la chromatine (Figure 3A). Nous avons proposé que cette propriété résulte d’une inhibition mutuelle entre l’activité des protéines Polycomb et la transcription. Lorsque la reprogrammation cellulaire nécessite l’activation de gènes sujets à des basculements cis-épigénétiques, l’outil CRISPRa est en principe préférable à une approche transgénique. En effet, les méthodes utilisant des transgènes exigent la présence continue (et donc potentiellement problématique) d’ADN transgénique, alors qu’une activation transitoire avec CRISPRa suffit à induire un changement durable de l’expression des gènes endogènes. Deux études d’une équipe de l’université de Duke, aux États-Unis, ont en effet démontré la supériorité d’une activation transcriptionnelle transitoire avec l’outil CRISPRa sur une approche de surexpression temporaire à partir d’un transgène. Dans les deux cas — génération de cellules neuronales à partir de fibroblastes embryonnaires [36] ou de cellules myogéniques à partir de cellules souches pluripotentes [37] — les auteurs ont démontré que seule une activation endogène permet un changement stable de l’expression des gènes de reprogrammation et une différenciation cellulaire efficace.
Modifications de la chromatine et mémoire épigénétique
Les approches d’édition de l’épigénome, de façon complémentaire aux analyses locales menées sur transgènes, permettent de tester la capacité des marques de la chromatine à stimuler leur propre maintien au cours des divisions cellulaires. Ces approches permettent de modifier transitoirement une région précise de la chromatine, puis d’évaluer si la machinerie cellulaire est capable de maintenir cette modification dans le temps. Bien qu’il s’agisse d’un domaine en cours d’exploration, les études menées jusqu’à présent suggèrent que la plupart des modifications individuelles n’ont pas, à elles seules, une capacité à stimuler leur propre maintien de façon stable au cours de divisions cellulaires. Il semble plutôt que le maintien d’un état chromatinien varie d’un type cellulaire à l’autre et dépende étroitement de la présence de plusieurs modifications de la chromatine et/ou de séquences d’ADN particulières. Par exemple, le maintien de la méthylation de la lysine en position 9 de l’histone H3 (H3K9me3) semble systématiquement corrélé à la présence de méthylation de l’ADN [38,39]. Les études réalisées jusqu’à présent suggèrent que les phénomènes de mémoire transcriptionnelle ne sont probablement pas réductibles aux propriétés épigénétiques de marques isolées.
Même si les modifications individuelles de la chromatine ne sont souvent pas suffisantes à induire un état épigénétiquement stable, elles peuvent, dans certains cas, être nécessaires au maintien d’un état chromatinien. Lorsque c’est le cas, un effacement transitoire d’une modification chromatinienne peut induire un changement durable de l’état chromatinien et de l’expression des gènes associés. Le recrutement ciblé d’une fusion de dCas9 avec le domaine catalytique de la déméthylase d’ADN Tet1 (ten-eleven translocation 1) au niveau du gène SNRPN (small nuclear ribonucleoprotein polypeptide N), qui est anormalement réprimé chez les personnes atteintes du syndrome de Prader-Willi1, permet, par exemple, l’effacement localisé de la méthylation de l’ADN et une réactivation stable de l’expression du gène dans des cellules en culture [40].
Implémentation artificielle d’un état chromatinien répressif
Les approches que nous avons décrites reposent sur la faculté inhérente des gènes manipulés à propager un changement d’expression, que ce soit par des mécanismes trans-épigénétiques ou cis-épigénétiques. Or, de nombreux gènes ne sont pas normalement dotés de tels mécanismes. Des outils, développés plus récemment, pourraient, dans certains cas, permettre de contourner cette limitation, en imposant à ces gènes une structure chromatinienne proche de celle présente au niveau d’éléments transposables ou de gènes soumis à empreinte parentale. Comme nous l’avons évoqué, le domaine KRAB de l’outil CRISPRi induit une hétérochromatinisation des gènes ciblés, caractérisée notamment par la déposition de la modification d’histone H3K9me3. Nous avons vu que le maintien de H3K9me3 dépend de la présence additionnelle de méthylation sur les cytosines de l’ADN. Mais, à elle seule, la méthylation de l’ADN ne suffit pas toujours à induire une répression stable. Il semble donc que la marque H3K9me3 et la méthylation de l’ADN agissent conjointement pour maintenir la répression transcriptionnelle. De façon remarquable, l’ajout d’un module de méthylation de l’ADN à la fusion dCas9-KRAB (outil baptisé CRISPR-off) permet d’induire une répression épigénétiquement stable au niveau de gènes qui ne sont pas normalement le siège d’un tel processus [41,42] (Figure 3B). Une équipe de l’hôpital San Raffaele de Milan, en Italie, a testé le principe d’une thérapie « épigénétique » fondée sur cette méthode [43]. Les auteurs de l’étude ont injecté à des souris des ARN messagers codant des éditeurs épigénétiques dirigés contre le gène Pcsk9 (proprotein convertase subtilisin/kexin Type 9)2, qui code une enzyme dont l’activité est augmentée chez des patients atteints d’hypercholestérolémie familiale. En utilisant des nanoparticules lipidiques comme vecteur des ARN messagers, ces auteurs ont réussi à cibler les cellules du foie et à réduire la production de la protéine PCSK9.
Un argument fréquent en faveur de l’édition épigénomique en tant qu’outil thérapeutique est que ses effets sont généralement plus faciles à inverser que ceux de l’édition du génome. Par exemple, Nuñez et al. [42] ont montré, par des expériences sur cellules en culture, qu’il est possible de réactiver l’expression d’un gène précédemment rendu silencieux. Pour y parvenir, ces auteurs ont fusionné le domaine Tet1 (Tet methylcytosine dioxygenase 1) au système CRISPRa afin d’effacer la méthylation de l’ADN et permettre une réactivation transcriptionnelle efficace. Ainsi, au système CRISPR-off de modification épigénétique correspond un système, baptisé CRISPR-on, capable de restaurer l’état épigénétique initial.
Vers des approches synthétiques d’édition de l’épigénome
Dans un effort de repousser les limites de l’édition du génome, une équipe de l’université de Boston, États-Unis, a mis au point un système artificiel de régulation épigénétique [44]. Ce système s’inspire du modèle dominant de propagation temporelle des états chromatiniens, selon lequel des systèmes enzymatiques de « lecture-écriture » participent au maintien des modifications de la chromatine après chaque division cellulaire. Les auteurs ont utilisé le domaine catalytique de l’enzyme bactérienne Dam qui catalyse la méthylation des adénines de l’ADN (m6A), une modification très peu abondante dans les génomes de mammifères. Ce premier module d’« écriture » permet, lorsqu’il est fusionné à un domaine programmable de liaison à l’ADN, de catalyser la déposition locale de la marque m6A au niveau d’un gène cible. Un second module de « lectureécriture » est composé de la fusion entre un domaine de liaison à la modification m6A (issu de la protéine bactérienne DpnI, qui agit en tant que module de « lecture ») et le domaine catalytique de l’enzyme Dam (qui fait office de module d’« écriture »). Ce module permet une propagation spatiale et temporelle de la modification m6A initialement déposée. Enfin, un troisième module « effecteur », composé de la fusion du module de « lecture » et d’un domaine d’activation transcriptionnelle, permet d’activer la transcription du gène cible en présence de la modification m6A. Bien que cette approche soit prometteuse, elle ne permet pas d’assurer une stabilité des changements d’expression équivalente à celle des méthodes mentionnées plus haut. De plus, ce type de système d’ingénierie artificielle de l’épigénome utilise des entités, comme le module de lecture-écriture et le module effecteur, qui ne sont pas naturellement présents dans les cellules de mammifères et qui doivent donc être exprimés de façon continue. Malgré ces défis pratiques, cette étude permet d’envisager des méthodes d’ingénierie épigénétique qui échappent aux contraintes des systèmes de régulation endogènes.
Conclusions et perspectives
Depuis plusieurs décennies, le génome n’est plus perçu comme un programme figé, mais comme la matrice sur laquelle se déploient des régulations complexes de l’expression génique. Les outils d’ingénierie de l’épigénome offrent, pour la première fois, la possibilité d’interférer directement avec ces processus de façon précise. Leurs contributions à la recherche biomédicale sont multiples et déterminantes.
Premièrement, en tant qu’outils, les approches de modification ciblée de l’épigénome contribuent à une meilleure compréhension des processus de régulation génique. Un enjeu majeur est notamment de déchiffrer les logiques régulatrices qui assurent la stabilité de l’expression génique au sein des différents types cellulaires. Comme nous l’avons vu, les modes de régulation déjà connus suggèrent que les systèmes épigénétiques font souvent intervenir plusieurs entités moléculaires dont la dynamique d’interaction permet la propagation temporelle d’un état d’expression génique. Les outils d’ingénierie de l’épigénome permettent de mieux étudier ces coopérations, car ils donnent la possibilité d’interférer localement avec différents processus moléculaires, individuellement ou conjointement.
Deuxièmement, grâce aux connaissances acquises sur les systèmes épigénétiques, les approches d’ingénierie ciblée de l’épigénome offrent de nouvelles perspectives thérapeutiques fondées sur la modulation de l’expression génique, que ce soit de manière transitoire ou durable. Le haut degré de précision des outils de modulation locale de l’épigénome, ainsi que la possibilité de contrôler temporellement les effets qu’ils produisent, ouvrent la voie à des applications potentiellement révolutionnaires, au moins aussi importantes que celles fondées sur la modification ciblée du génome.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Le syndrome de Prader-Willi est un trouble du développement lié à un défaut d’expression de gènes de la région 15q11-q12.
La PCSK9 dégrade les récepteurs qui captent les particules de cholestérol présentes dans le sang et les éliminent de la circulation, conduisant à une accumulation de cholestérol.
Références
- Blau HM, Daley GQ. Stem Cells in the Treatment of Disease. N Engl J Med 2019 ; 380 : 1748–60. [CrossRef] [PubMed] [Google Scholar]
- Atlasi Y, Stunnenberg HG. The interplay of epigenetic marks during stem cell differentiation and development. Nat Rev Genet 2017 ; 18 : 643–58. [CrossRef] [PubMed] [Google Scholar]
- Luger K, Mäder AW, Richmond RK, et al. Crystal structure of the nucleosome core particle at 2,8 Å resolution. Nature 1997 ; 389 : 251–60. [CrossRef] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science 2001 ; 293 : 1074–80. [CrossRef] [PubMed] [Google Scholar]
- Kadonaga JT. Regulation of RNA polymerase II transcription by sequence-specific DNA binding factors. Cell 2004 ; 116 : 247–57. [CrossRef] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell 2007 ; 128 : 707–19. [CrossRef] [PubMed] [Google Scholar]
- Teves SS, Weber CM, Henikoff S. Transcribing through the nucleosome. Trends Biochem Sci 2014 ; 39 : 577–86. [CrossRef] [PubMed] [Google Scholar]
- Mehrmohamadi M, Sepehri MH, Nazer N, et al. A Comparative Overview of Epigenomic Profiling Methods. Front Cell Dev Biol 2021 ; 9 : 714687. [CrossRef] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993 ; 118 : 401–15. [CrossRef] [PubMed] [Google Scholar]
- Bryant GO, Ptashne M. Independent recruitment in vivo by Gal4 of two complexes required for transcription. Mol Cell 2003 ; 11 : 1301–9. [CrossRef] [PubMed] [Google Scholar]
- Sgro A, Blancafort P. Epigenome engineering: new technologies for precision medicine. Nucleic Acids Res 2020 ; 48 : 12453–82. [CrossRef] [PubMed] [Google Scholar]
- Nakamura M, Gao Y, Dominguez AA, et al. CRISPR technologies for precise epigenome editing. Nat Cell Biol 2021 ; 23 : 11–22. [CrossRef] [PubMed] [Google Scholar]
- Desjarlais JR, Berg JM. Use of a zinc-finger consensus sequence framework and specificity rules to design specific DNA binding proteins. Proc Natl Acad Sci U S A 1993 ; 90 : 2256–60. [CrossRef] [PubMed] [Google Scholar]
- Bibikova M, Golic M, Golic KG, et al. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 2002 ; 161 : 1169–75. [CrossRef] [PubMed] [Google Scholar]
- Beerli RR, Segal DJ, Dreier B, et al. Toward controlling gene expression at will: Specific regulation of the erbB - 2 / HER - 2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc Natl Acad Sci USA 1998 ; 95 : 14628–33. [CrossRef] [PubMed] [Google Scholar]
- Snowden AW, Gregory PD, Case CC, et al. Gene-specific targeting of H3K9 methylation is sufficient for initiating repression in vivo. Curr Biol 2002 ; 12 : 2159–66. [CrossRef] [PubMed] [Google Scholar]
- Carvin CD, Parr RD, Kladde MP. Site-selective in vivo targeting of cytosine-5 DNA methylation by zinc-finger proteins. Nucleic Acids Res 2003 ; 31 : 6493–501. [CrossRef] [PubMed] [Google Scholar]
- Kay S, Hahn S, Marois E, et al. A bacterial effector acts as a plant transcription factor and induces a cell size regulator. Science 2007 ; 318 : 648–51. [CrossRef] [PubMed] [Google Scholar]
- Bogdanove AJ, Schornack S, Lahaye T. TAL effectors: finding plant genes for disease and defense. Curr Opin Plant Biol 2010 ; 13 : 394–401. [CrossRef] [PubMed] [Google Scholar]
- Becker S, Boch J. TALE and TALEN genome editing technologies. Gene and Genome Editing 2021 ; 2 : 100007. [CrossRef] [Google Scholar]
- Nitsch S, Mussolino C. Generation of TALE-Based Designer Epigenome Modifiers. Methods Mol Biol 2018 ; 1767 : 89–109. [CrossRef] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014 ; 346 : 1258096. [CrossRef] [PubMed] [Google Scholar]
- Liu Z, Dong H, Cui Y, et al. Application of different types of CRISPR/Cas-based systems in bacteria. Microbial Cell Factories 2020 ; 19 : 172. [CrossRef] [PubMed] [Google Scholar]
- Larson MH, Gilbert LA, Wang X, et al. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc 2013 ; 8 : 2180–96. [CrossRef] [PubMed] [Google Scholar]
- Maeder ML, Linder SJ, Cascio VM, et al. CRISPR RNA-guided activation of endogenous human genes. Nat Methods 2013 ; 10 : 977–9. [CrossRef] [PubMed] [Google Scholar]
- Chavez A, Tuttle M, Pruitt BW, et al. Comparison of Cas9 activators in multiple species. Nat Methods 2016 ; 13 : 563–7. [CrossRef] [PubMed] [Google Scholar]
- Thakore PI, D’Ippolito AM, Song L, et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods 2015 ; 12 : 1143–9. [CrossRef] [PubMed] [Google Scholar]
- Fulco CP, Munschauer M, Anyoha R, et al. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 2016 ; 354 : 769–73. [CrossRef] [PubMed] [Google Scholar]
- Hilton IB, D’Ippolito AM, Vockley CM, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 2015 ; 33 : 510–7. [CrossRef] [PubMed] [Google Scholar]
- Wang K, Escobar M, Li J, et al. Systematic comparison of CRISPR-based transcriptional activators uncovers gene-regulatory features of enhancer-promoter interactions. Nucleic Acids Res 2022 ; 50 : 7842–55. [CrossRef] [PubMed] [Google Scholar]
- Braun SMG, Kirkland JG, Chory EJ, et al. Rapid and reversible epigenome editing by endogenous chromatin regulators. Nat Commun 2017 ; 8 : 560. [CrossRef] [PubMed] [Google Scholar]
- Tanenbaum ME, Gilbert LA, Qi LS, et al. A Protein-Tagging System for Signal Amplification in Gene Expression and Fluorescence Imaging. Cell 2014 ; 159 : 635–46. [CrossRef] [PubMed] [Google Scholar]
- Policarpi C, Munafò M, Tsagkris S, et al. Systematic epigenome editing captures the context-dependent instructive function of chromatin modifications. Nat Genet 2024 ; 56 : 1168–80. [CrossRef] [PubMed] [Google Scholar]
- Chakraborty S, Ji H, Kabadi AM, et al. A CRISPR/Cas9-Based System for Reprogramming Cell Lineage Specification. Stem Cell Rep 2014 ; 3 : 940–7. [CrossRef] [Google Scholar]
- Holoch D, Wassef M, Lövkvist C, et al. A cis-acting mechanism mediates transcriptional memory at Polycomb target genes in mammals. Nat Genet 2021 ; 53 : 1686–97. [CrossRef] [PubMed] [Google Scholar]
- Black JB, Adler AF, Wang H-G, et al. Targeted Epigenetic Remodeling of Endogenous Loci by CRISPR/Cas9-Based Transcriptional Activators Directly Converts Fibroblasts to Neuronal Cells. Cell Stem Cell 2016 ; 19 : 406–14. [CrossRef] [PubMed] [Google Scholar]
- Kwon JB, Vankara A, Ettyreddy AR, et al. Myogenic Progenitor Cell Lineage Specification by CRISPR/Cas9-Based Transcriptional Activators. Stem Cell Rep 2020 ; 14 : 755–69. [CrossRef] [Google Scholar]
- Carlini V, Policarpi C, Hackett JA. Epigenetic inheritance is gated by naïve pluripotency and Dppa2. EMBO J 2022 ; 41 : e108677. [CrossRef] [PubMed] [Google Scholar]
- Hathaway NA, Bell O, Hodges C, et al. Dynamics and Memory of Heterochromatin in Living Cells. Cell 2012 ; 149 : 1447–60. [CrossRef] [PubMed] [Google Scholar]
- Rohm D, Black JB, McCutcheon SR, et al. Activation of the imprinted Prader-Willi Syndrome locus by CRISPR-based epigenome editing. bioRxiv 2024 Mar 4:2024.03.03.583177. [PubMed] [Google Scholar]
- Amabile A, Migliara A, Capasso P, et al. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016 ; 167 : 219–32.e14. [CrossRef] [PubMed] [Google Scholar]
- Nuñez JK, Chen J, Pommier GC, et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 2021 ; S0092867421003536. [Google Scholar]
- Cappelluti MA, Mollica Poeta V, Valsoni S, et al. Durable and efficient gene silencing in vivo by hit-and-run epigenome editing. Nature 2024 ; 627 : 416–23. [CrossRef] [PubMed] [Google Scholar]
- Park M, Patel N, Keung AJ, et al. Engineering Epigenetic Regulation Using Synthetic Read-Write Modules. Cell 2019 ; 176 : 227–38.e20. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1 Les principales familles d’outils d’ingénierie épigénétique. A. Zinc Fingers (ZF) – Les domaines ZF reconnaissent une séquence d’ADN spécifique et permettent d’y cibler un domaine effecteur. B. TALE – Le domaine effecteur est lié au domaine de reconnaissance et de liaison à l’ADN composé d’arrangements variables de modules liant chacun un des quatre nucléotides de l’ADN. C. Système CRISPR-dCas9 – L’ARN guide s’apparie à la séquence cible et permet l’amarrage de la protéine dCas9 fusionnée au domaine effecteur. |
| Dans le texte | |
|
Figure 2 Outils d’édition de l’épigénome basés sur l’approche CRISPR/dCas9. A. La méthode « CRISPR interference » (CRISPRi) utilise une fusion de la version inactive de Cas9 (dCas9) avec le domaine KRAB, lequel induit la répression transcriptionnelle du gène cible en établissant un état chromatinien répressif. B. Le système « CRISPR activation » (CRISPRa) résulte de la fusion dCas9 avec un ou plusieurs activateurs transcriptionnels. Il permet d’activer l’expression du gène cible grâce à l’établissement d’un état chromatinien permissif et au recrutement de la machinerie transcriptionnelle. C. La fusion de différents domaines effecteurs de régulateurs épigénétiques à la protéine dCas9 permet de moduler la composition de la chromatine via l’ajout ou l’effacement de diverses modifications chimiques, ainsi que par l’ajout ou l’éjection de nucléosomes. HMT : histone méthyltransférase ; HAT : histone acétyltransférase ; HDAC : histone déacétylase ; KDM : lysine déméthylase. |
| Dans le texte | |
|
Figure 3 Les différentes approches d’édition de l’épigénome qui permettent d’induire des changements stables d’expression génique. A. Une activation transitoire peut être maintenue de façon prolongée si le gène est soumis à un processus de mémoire trans-épigénétique (comme c’est le cas lorsque le produit du gène activé est capable de maintenir sa propre transcription) ou à un processus de mémoire cis-épigénétique, dont la logique repose sur une opposition mutuelle entre les protéines Polycomb et la transcription. B. Le recrutement transitoire d’une protéine de fusion entre dCas9, le domaine KRAB et un module de méthylation de l’ADN permet d’instaurer une répression cis-épigénétique semblable à celle existant au niveau des éléments transposables ou de certains gènes soumis à une empreinte. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.