")
")
| Issue |
Med Sci (Paris)
Volume 40, Number 12, Décembre 2024
Épigénétique : développement et destin cellulaire
|
|
|---|---|---|
| Page(s) | 925 - 934 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2024180 | |
| Published online | 20 December 2024 | |
Épigénétique et cancer
La méthylation dans tous ses états
Epigenetics and cancer: the role of DNA methylation
Université de Rennes, Inserm, EHESP, Irset (Institut de recherche en santé, environnement et travail) UMR_S 1085, Rennes, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les altérations des profils de méthylation de l’ADN sont retrouvées dans les cellules cancéreuses, combinant une hypométhylation globale du génome avec une hyperméthylation de régions spécifiques, telles que les îlots CpG, normalement non méthylés. Des effets moteurs dans le développement du cancer ont été associés à certaines régions modifiées par la méthylation de l’ADN, induisant par exemple la répression de gènes suppresseurs de tumeur ou l’activation d’oncogènes et de rétrotransposons. Ces altérations représentent des candidats de premier plan pour le développement de marqueurs spécifiques pour la détection, le diagnostic et le pronostic du cancer. En particulier, ces marqueurs, distribués le long du génome, représentent une mine d’informations qui offre des perspectives d’innovation en biopsie liquide, notamment grâce à l’émergence de l’intelligence artificielle à visée diagnostique. Ceci pourrait lever les verrous liés aux sensibilités et spécificités qui restent encore limitées pour les applications les plus difficiles en oncologie : la détection des cancers à un stade précoce, le suivi de la maladie résiduelle et l’analyse des tumeurs cérébrales. Le ciblage des processus enzymatiques qui contrôlent l’épigénome offrent en outre de nouvelles stratégies thérapeutiques qui pourraient remédier aux anomalies de régulation de ces épigénomes altérés.
Abstract
Alterations in DNA methylation profiles are typically found in cancer cells, combining genome-wide hypomethylation with hypermethylation of specific regions, such as CpG islands, which are normally unmethylated. Driving effects in cancer development have been associated with alteration of DNA methylation in certain regions, inducing, for example, the repression of tumor suppressor genes or the activation of oncogenes and retrotransposons. These alterations represent prime candidates for the development of specific markers for the detection, diagnosis and prognosis of cancer. In particular, these markers, distributed along the genome, provide a wealth of information that offers potential for innovation in the field of liquid biopsy, in particular thanks to the emergence of artificial intelligence for diagnostic purposes. This could overcome the limitations related to sensitivities and specificities, which remain too low for the most difficult applications in oncology: the detection of cancers at an early stage, the monitoring of residual disease and the analysis of brain tumors. In addition, targeting the enzymatic processes that control the epigenome offers new therapeutic strategies that could reverse the regulatory anomalies of these altered epigenomes.
© 2024 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (© Biorender.com).
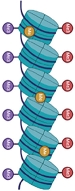
De nombreuses études ont révélé le rôle central des processus é pigé né tiques dans les causes, l’évolution et le traitement des cancers [1,2]. La reprogrammation épigénétique est en effet une des caractéristiques distinctives des génomes cancéreux, facilitant l’acquisition de capacités tumorales [3]. Les perturbations des marques épigénétiques peuvent induire un cancer, indépendamment de toute mutation [4]. En particulier, les profils aberrants de la méthylation de l’ADN sont une des grandes caractéristiques des cellules néoplasiques [1]. La méthylation de l’ADN est le premier mécanisme épigénétique qui a été décrit en lien avec la répression transcriptionnelle et la différenciation cellulaire [5,6], et qui est considérée comme un élément clé de la régulation épigénétique. Contrairement aux modifications d’histones, qui fournissent un système flexible pour la mise en place et l’effacement de certains états transcriptionnels, la méthylation de l’ADN induit une répression à long terme et sert souvent de mécanisme de fixation d’un état réprimé, tel un cadenas [7]. Chez les mammifères, la méthylation de l’ADN modifie principalement les cytosines (C) des dinucléotides CpG. Cette marque épigénétique est enrichie dans les séquences pauvres en CpG. En revanche, environ 95 % des séquences riches en CpG, également connues sous le nom d’îlots CpG, ne sont pas méthylées (Figure 1A) [6,8] (→).
(→) Voir la Synthèse de M. Weber, m/s n° 8-9, aoûtseptembre 2008, page 731
 |
Figure 1 Profils de méthylation différentiels caractéristiques des génomes tumoraux. A. Les génomes somatiques sains sont globalement méthylés, excepté au niveau des îlots CpG : la vaste majorité des îlots CpG n’est pas méthylée dans les génomes sains. Ceci concerne également 72 % des promoteurs associés à un îlots CpG, quel que soit le statut d’expression du gène régulé par ce promoteur [6]. B. Les génomes tumoraux combinent hypométhylation et hyperméthylation acquises au cours de la carcinogenèse (illustration conçue avec BioRender.com). |
De nombreux promoteurs (72 %) sont associés à ces îlots CpG majoritairement non méthylés, indépendamment du statut d’expression des gènes qu’ils régulent [6]. La méthylation est distribuée sur une grande partie des génomes mammifères, couvrant environ 80 % des CpG dans les cellules somatiques [9]. On parle de « génomes globalement méthylés ». Par défaut, le terme de méthylation de l’ADN se réfère à la 5-méthylcytosine (5mC), la base modifiée la plus représentée dans ces génomes [9]. Plus récemment, la mise en évidence de la 5-hydroxyméthylcytosine (5hmC), intermédiaire de déméthylation dérivé de la 5mC, a aussi été étudiée dans les processus de développement du cancer. Cette dernière est cependant largement moins abondante, couvrant moins d’1 % des cytosines du génome (entre 0,03 et 0,7 % selon les contextes) contre environ 5 % des cytosines modifiées en 5mC [10,11].
Chez les mammifères, les profils de méthylation des génomes sont mis en place de novo dans la lignée germinale et au cours du développement embryonnaire précoce, puis maintenus par un mécanisme de reproduction fidèle sur chaque brin néosynthétisé au cours de la division cellulaire. La famille des ADN méthyltransférases, enzymes responsables de l’établissement et du maintien de ces marques, est composée de quatre membres, regroupés en deux familles structurellement et fonctionnellement distinctes : DNMT1, l’ADN méthyltransférase de maintenance, et le groupe des DNMT3, les ADN méthyltransférases de novo et leur cofacteur [12]. La déméthylation active de l’ADN, par retrait des groupes méthyle, est effectuée par les enzymes de la famille TET (ten-eleven translocation) [11].
Les mécanismes d’altération de la méthylation de l’ADN dans le cancer
L’altération des profils de méthylation de l’ADN est l’un des changements moléculaires les plus précoces et les plus systématiques des tumeurs humaines [1,13]. L’hyperméthylation d’un large éventail de régions spécifiques telles que les îlots CpG associés aux promoteurs des gènes suppresseurs de tumeurs [14], parallèlement à une perte de méthylation globale du génome [15] (Figure 1B) sont classiquement observés. Ces altérations peuvent résulter de plusieurs mécanismes.
Mutations dans la machinerie de méthylation et de déméthylation de l’ADN
Des mutations somatiques dans les gènes des ADN méthyltransférases ont été liées à la transformation maligne des cellules [16]. L’inactivation de l’enzyme de maintenance DNMT1 peut entraîner une perte globale de méthylation de l’ADN, alors que l’expression ectopique des méthylases de novo DNMT3 peut être à l’origine de l’établissement de marques de méthylation aberrantes, telles que l’hyperméthylation des îlots CpG. Cependant, les mutations des DNMT chez l’homme sont étonnamment rares et ne semblent couvrir qu’une petite fraction des cancers [12]. La recherche sur le rôle des DNMT dans le développement des tumeurs repose, actuellement, sur quelques modèles murins [8,12] et l’impact fonctionnel de chaque enzyme reste à préciser. En revanche, les gènes codant les enzymes TET, en particulier TET2, sont fréquemment mutés dans divers cancers. Des mutations faux-sens ou aboutissant à une protéine tronquée sont observées dans presque tous les types de tumeurs, bien qu’à une fréquence relativement faible (0,1 % à 10 % des cas). Dans certaines études jusqu’à 20 % des patients, par exemple dans le mélanome et le cancer colorectal, sont porteurs de mutations dans un ou plusieurs gènes TET [11]. Cependant, il reste difficile de déterminer si les mutations de TET confèrent un avantage sélectif ou représentent des altérations passagères sans effets directs sur la carcinogénèse.
Les autres mutations de la machinerie d’établissement de l’épigénome
Les méthodes de séquençage de l’exome (les portions codantes du génome) ont précédemment révélé une fréquence élevée de mutations dans des gènes connus pour participer directement à l’organisation de l’épigénome, et ce dans de nombreux types de tumeurs [1,16]. Nombre de ces mutations, couvrant d’autres gènes que ceux codant les enzymes DNMT et TET, aboutissent également à des profils de méthylation aberrants. Une étude, comparant plus de 6 000 méthylomes (profils de méthylation) issus de différents types de cancers, a montré que les tumeurs présentant des mutations des facteurs de remodelage de la chromatine et de la signalisation Wnt étaient affectées par une instabilité de la méthylation de l’ADN, caractérisée par de nombreux locus hyper- et hypo-méthylés [17].
On peut citer, par exemple, les mutations gain de fonction du répresseur Polycomb (PRC) EZH2 (enhancer of zeste homolog 2), qui favorisent la restriction de la chromatine et entravent la différenciation par une activité répressive excessive [2]. Or, il a été montré que les gènes qui gagnent de la méthylation de l’ADN dans les cancers sont fortement biaisés en faveur des gènes normalement marqués par la tri-méthylation de la lysine 27 de la protéine histone H3 (H3K27me3), marque déposée par EZH2, dans les cellules souches embryonnaires et adultes [16]. Une rétention anormale des complexes PRC peut se produire dans un premier temps, suivie d’une méthylation de l’ADN. Les protéines PRC semblent servir de plateformes de recrutement pour les DNMT impliquées dans l’hyperméthylation des gènes suppresseurs de tumeurs, ce qui met en évidence un autre lien entre divers systèmes de répression épigénétique [18]. Le niveau d’expression des gènes dans le tissu sain détermine leur propension à être hyperméthylés dans les cellules cancéreuses. Il a d’ailleurs été montré que la méthylation aberrante de l’ADN peut servir de marqueur du lignage cellulaire [19]. Comme dans le cancer de la prostate, cela concerne des gènes « prémarqués » par H3K27me3, mais pas seulement [20]. Ceci pose la question de l’état transcriptionnel et du possible « pré-marquage » dans les tissus sains ou précancéreux des gènes suppresseurs de tumeur retrouvés hyperméthylés dans les cancers.
Des mutations dans les gènes métaboliques IDH1/2 (isocitrate dehydrogenase 1/2), SDH (succinate dehydrogenase) et FH (fumarate hydratase) sont également retrouvées dans une grande variété de tumeurs solides et sont suspectées d’affecter les profils de méthylation de l’ADN via l’inhibition de l’expression des enzymes TET [1,11], aboutissant notamment à une hyperméthylation du génome. Enfin, d’autres études ont révélé que des facteurs interagissant avec TET2, telles que les protéines IDAX/CXXC4 (inhibitor of disheveled and axin / CXXC finger protein 4) et RINF/CXXC5 (retinoid-inducible nuclear factor / CXXC finger protein 5) ou certains micro-ARN, comme miR-22, sont surexprimés dans plusieurs tumeurs solides, y compris le cancer du sein et le cancer colorectal, et auraient un impact négatif sur la stabilité et la fonction de la protéine TET2 [11]. La régulation à la baisse de la famille des micro-ARN miRNA-29, qui a été reliée à la surexpression des ADN méthyltransférases, est un autre exemple [1].
Les autres mécanismes : l’impact de l’environnement tumoral
L’hypométhylation globale du génome pourrait en partie résulter d’un taux lent de reméthylation de l’ADN après la réplication, à l’origine d’une restauration incomplète du méthylome lors de la mitose [21]. En effet, les régions hypométhylées dans le cancer couvrent particulièrement les domaines de réplication tardive [14,15,22]. La lenteur de reproduction des profils de méthylation par la machinerie de maintenance pourrait donc être à l’origine d’une perte passive de méthylation au cours des divisions cellulaires.
Certaines études montrent l’existence de liens entre le métabolisme des folates et l’activité des méthylases, le rôle des facteurs environnementaux qui favorisent l’hyperméthylation de l’ADN dans les tissus gastrointestinaux, et les effets potentiels du stress micro-environnemental sur l’expression des régulateurs de la chromatine [2]. Par exemple, l’hypoxie est connue pour réduire l’activité des déméthylases TET, ce qui entraîne des changements importants dans le méthylome, dont une hyperméthylation [3]. Ces observations suggèrent ainsi qu’une combinaison de mutations affectant les enzymes directement responsables de l’établissement des profils de méthylation de l’ADN, mais surtout des altérations de leurs régulateurs et un taux de prolifération élevé peuvent provoquer les modifications des profils des 5mC et 5hmC.
Les conséquences des altérations, cinétique, effets moteurs ou secondaires
Les altérations du méthylome entraînent la reprogrammation transcriptomique des génomes cancéreux en reproduisant des contextes similaires à des mutations oncogéniques : inhibition de gènes suppresseurs de tumeurs et/ou activation d’oncogènes présentant des effets moteurs évidents sur la carcinogénèse. L’étude des 6 000 méthylomes que nous avons citée plus haut [17] a identifié 581 gènes réprimés et 85 gènes surexprimés en lien avec des statuts de méthylation spécifiques du génome de cellules cancéreuses. La plupart de ces gènes réprimés ou surexprimés sont impliqués dans des voies de signalisation spécifiques, incluant l’apoptose, les voies de réparation de l’ADN et le métabolisme cellulaire. Ceci indique un rôle plus global des changements de méthylation dans la transformation cellulaire.
L’hyperméthylation des îlots CpG
Le phénotype d’hyperméthylation des îlots CpG (CIMP), localisés au niveau de promoteurs de gènes suppresseurs de tumeurs, est associé à la répression de ces gènes (Figure 1) et peut clairement contribuer à la transformation cellulaire [18]. Les exemples les plus documentés touchent notamment les gènes MLH11, BRCA12, MGMT3, APC4, RB15, VHL6, CDKN2A7 [1,17]. Cependant, le rôle généralisé de cette hyperméthylation dans la répression des gènes associés au cancer reste questionné. Il a, par exemple, été montré dans les gliomes que les altérations de H3K27me3 représentent le défaut moléculaire principal au niveau des gènes dérégulés transcriptionnellement [23]. La méthylation de l’ADN des régions promotrices de certains gènes codant des micro-ARN peut aussi modifier leurs profils d’expression et, indirectement, l’expression de leurs cibles [18]. L’hyperméthylation de régions spécifiques a été associée à une différenciation cellulaire incomplète [24] et à l’acquisition d’un phénotype « souche », établissant un lien direct entre les modifications de la méthylation de l’ADN et la transformation oncogène. Les cellules cancéreuses peuvent devenir dépendantes de leur profil aberrant de méthylation de l’ADN, en particulier de l’hyperméthylation, qui réprime des gènes impliqués dans des voies de signalisation oncogènes mais aussi des voies de signalisation non-oncogènes qui sont requises pour leur survie [25]. En raison de ces circuits aberrants, les cellules cancéreuses deviennent hypersensibles aux effets des gènes suppresseurs de tumeurs classiques [26] mais aussi aux gènes qui peuvent inhiber les voies de signalisation non-oncogéniques sur lesquelles elles s’appuient pour survivre [25]. Identifier ces cibles pourrait permettre le développement de thérapies épigénétiques ciblées. D’autre part, l’hyperméthylation, à des stades avancés de la carcinogenèse, semble jouer un rôle protecteur contre la progression du cancer en limitant la plasticité épigénétique et le potentiel adaptatif [23].
L’hypométhylation des régions riches en méthylation
L’hypométhylation de régions normalement méthylées peut entraîner la réactivation des éléments sous le contrôle de cette méthylation (Figure 1). C’est le cas de certains oncogènes, comme MYCN8, BCL2L109, CTNNB110, IRS211 et IGF212 [17] et de la famille des gènes cancer-testis specific, tels que MAGEC213 et SYCP214, normalement exprimés exclusivement dans les cellules germinales, en particulier les cellules mâles [8,27] (→).
(→) Voir la Synthèse de S. Rousseaux et al., m/s n° 8-9, août-septembre 2008, page 735
Ces gènes anormalement réactivés, sont alors responsables de la production d’antigènes spécifiques de la tumeur, et présentent un intérêt particulier comme biomarqueurs et cibles thérapeutiques.
L’hypométhylation globale observée dans les génomes tumoraux est principalement retrouvée dans les régions intergéniques, en particulier celles enrichies en éléments répétés transposables du type Alu15 et LINE-1 (L1, long interspersed nuclear elements-1) [28]. En effet, une hypométhylation a été rapportée dans presque toutes les classes d’éléments répétés et dans plusieurs formes de cancers [29]. Cela induit notamment la réactivation des éléments mobiles, les rétrotransposons, avec pour conséquence l’acquisition d’une instabilité génomique, des réarrangements chromosomiques et la production de transcrits chimériques entre l’élément transposable et son locus adjacent. L’hypométhylation du promoteur interne de LINE-1 (L1) a ainsi été décrit comme une caractéristique du cancer [29,30] qui peut avoir pour conséquence la réactivation des éléments L1 intacts [31]. Ceci aboutit à la production anormale de leurs transcrits et de leurs protéines. Une étude récente décrit la détection de la protéine de L1 ORF1p16 dans la circulation sanguine de patients atteints de plusieurs types de cancers [32], démontrant le potentiel d’utilisation des éléments L1 comme biomarqueurs non invasifs et multi-cancer. La rétrotransposition de ces éléments compétents induit des cassures doubles brins de l’ADN et endommage le génome. Ces dommages liés aux rétrotranspositions des éléments L1 ont été décrits comme particulièrement abondants dans quatre types de cancers (œsophage, tête et cou, poumon et colorectal) [33]. Par ailleurs, un évènement de transposition mutagène dans le gène APC (adenomatous polyposis coli) a été démontré comme responsable de l’initiation d’un cancer colorectal [34]. Selon certaines études, les changements épigénétiques pourraient être les premiers facteurs d’initiation d’un cancer chez l’homme [35]. En effet, les altérations de la méthylation de l’ADN ont été identifiées comme un évènement précoce de la transition des cellules normales en cellules cancéreuses [36]. Cependant, les modifications génétiques et épigénétiques sont inextricablement liées dans la génération des phénotypes malins. Il reste donc difficile de déterminer précisément les effets directs et indirects ainsi que la chronologie d’apparition des différents types d’altérations de la méthylation de l’ADN au cours du développement des tumeurs. Néanmoins, la méthylation différentielle caractéristique des génomes tumoraux paraît être un bon marqueur pour détecter le cancer dès les premiers stades de la maladie.
Le ciblage des altérations : voies diagnostiques et voies thérapeutiques
Les méthylomes aberrants fournissent des cibles thérapeutiques prometteuses, et comportent un niveau supplémentaire d’information par rapport à des méthodes interrogeant uniquement des altérations génétiques (Figure 2). Contrairement aux mutations ponctuelles, qui n’affectent qu’une unique paire de bases par génome, les altérations de la méthylation sont dispersées sur l’intégralité du génome et affectent de multiples résidus par région. La concordance de méthylation entre plusieurs sites CpG d’une même région peut permettre de détecter des anomalies de basse fréquence parmi une population hétérogène de molécules [37]. De plus, combiner plusieurs régions altérées permet de capturer une large gamme d’allèles tumoraux et ainsi de couvrir les profils hétérogènes des patients atteints de cancer. Les profils de méthylation de l’ADN peuvent aussi informer sur le tissu d’origine des tumeurs [1], ce qui n’est pas le cas des mutations oncogéniques qui sont communes à plusieurs types de cancers. Ainsi, de nouvelles stratégies de diagnostic intégrant des biomarqueurs é pigé né tiques, tels que la méthylation de l’ADN, permettent d’atteindre une haute sensibilité de détection et de couvrir les cas sans mutation.
 |
Figure 2 Défis et innovations en matière de détection des altérations tumorales à visée diagnostique : le potentiel de la méthylation de l’ADN circulant. Les progrès réalisés dans le domaine de la biopsie liquide ont démontré l’intérêt de quantifier l’ADN circulant tumoral (ADNct) pour améliorer la prise en charge des patients. Cependant, détecter les faibles fractions d’ADN tumoral, libérées aux stades précoces ou au cours du traitement, reste un défi et nécessite le développement de méthodes de détection et de quantification plus sensibles (illustration conçue avec BioRender.com). |
Les tumeurs solides libèrent, dans la circulation sanguine, du matériel tumoral tel que des cellules, des vésicules extracellulaires ou encore des fragments d’ADN. Des techniques permettent d’étudier les profils de méthylation de ces fragments d’ADN tumoraux dans divers types de cancers. Ces marqueurs, évalués par des processus non-invasifs, pourraient se révéler décisifs pour détecter les stades précoces de la maladie, lorsque les chances de guérison sont les meilleures, ou encore pour identifier la maladie résiduelle, les premières étapes de la rechute ou d’acquisition de la résistance au cours du traitement [38]. Ces approches promettent une gestion optimale des patients atteints de cancer et prennent actuellement une place importante en oncologie.
Biopsie liquide et méthylation de l’ADN circulant
Des recherches approfondies ont montré que les altérations génétiques tumorales peuvent être détectées dans le plasma des patients atteints de cancer, à partir de l’ADN qui en est extrait [39]. Cette possibilité de détection a ouvert la voie aux analyses moléculaires réalisées à partir de biopsies liquides pour déterminer le profil moléculaire des tumeurs, établir le pronostic et procéder à la surveillance de la maladie de manière non invasive.
L’ADN plasmatique, ou ADN circulant, est présent dans le sang sous la forme de fragments libres. Chez les patients atteints de cancer, une fraction de cet ADN plasmatique est issue de la tumeur (Figure 3). Cet ADN circulant tumoral est libéré par les cellules cancéreuses, lors du renouvellement cellulaire, au cours de la croissance tumorale [40]. Il porte donc les altérations moléculaires de la tumeur qui permettent de distinguer les fragments tumoraux des fragments sains, qui sont eux majoritairement issus des cellules hé matopoï é tiques [41,42]. De nombreuses études ont fait état de l’excellente concordance entre les altérations détectées dans l’ADN circulant tumoral et le tissu tumoral d’origine [43], démontrant son potentiel comme marqueur de la progression du cancer.
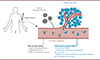 |
Figure 3 Biopsies liquides et ADN tumoral circulant (ADNct). L’ADN circulant (ADNc) est présent dans le système sanguin sous forme de fragments libres, qui peuvent être isolés à partir du plasma (la phase acellulaire du sang). Ces fragments présentent une taille moyenne de 167 paires de bases (bp), équivalente à la taille d’un brin d’ADN enroulé autour d’un nucléosome, ce qui suggère qu’ils sont majoritairement générés au cours de la dégradation du génome lors de la mort cellulaire par apoptose. Chez les patients atteints de cancer, une fraction de cet ADN plasmatique provient de la tumeur (illustration conçue avec BioRender.com). |
L’analyse de la méthylation de l’ADN à partir d’ADN circulant peut également contribuer à la détection, au pronostic, au sous-typage moléculaire et au suivi des patients atteints de cancer [44]. Les profils de méthylation de l’ADN cellulaire sont conservés dans les fragments d’ADN circulant, et il est possible de détecter des profils spécifiques de cancers à l’échelle du génome entier (voir [44]). Initialement, les études portant sur les profils de méthylation de l’ADN plasmatique ont ciblé un nombre limité de régions à une profondeur élevée, en utilisant des méthodes fondées sur la PCR (polymerase chain reaction), ou ont exploré l’ensemble du génome à une faible profondeur, grâce au séquençage à haut débit [44]. Plus récemment, des études s’appuyant sur la capture de régions d’intérêt par hybridation couplée au séquençage haut débit ont examiné les performances d’un grand nombre de régions à une profondeur élevée [37,45–51]. Ces approches ont permis une détection et une classification sensibles d’échantillons cancéreux à partir de l’ADN plasmatique. Toutefois, comme elles se concentrent essentiellement sur l’hyperméthylation et les séquences uniques, elles impliquent de cibler des régions spécifiques pour chaque sous-type de cancer. Pour dépasser cette limite, notre équipe a mis au point une méthode (DIAMOND, pour detection of long interspersed nuclear element altered methylation on plasma DNA), détectant les altérations de méthylation des rétrotransposons LINE-1 spécifiques des génomes de primates (type L1PA) à partir d’ADN circulant [52]. Nous avons ainsi établi que le ciblage de l’hypométhylation de ces rétrotransposons est un biomarqueur sensible et spécifique qui permet de détecter de multiples formes de cancer, et cela de manière non invasive. L’hypométhylation des éléments L1, qui est une caractéristique commune à de multiples formes de cancer, permet de couvrir, en un seul test, les profils hétérogènes des patients atteints de cancer. Cette approche offre un équilibre entre le nombre de régions ciblées et la profondeur de séquençage, ce qui pourrait améliorer considérablement la sensibilité de la détection de l’ADN circulant tumoral et améliorer la prise en charge des patients atteints d’un cancer. Notons que l’exploration fine des profils de méthylation de l’ADN circulant, fournissant une multitude d’informations, nécessitent le développement de modèles statistiques permettant d’extraire les signaux cancer-spécifiques (voir plus loin).
Voies thérapeutiques
Les acteurs moléculaires participant aux voies épigénétiques se sont révélés être des cibles thérapeutiques prometteuses. Les premières molécules testées pour interférer avec ces voies, la 5-azacytidine et la 2′-désoxy-5-azacytidine, sont des inhibiteurs de DNMT, et ont été approuvées pour le traitement des tumeurs malignes hématopoïétiques [53]. L’action de ces molécules consiste surtout à restaurer l’expression de gènes, dont l’extinction anormale contribue à la cancérogenèse. Ce traitement peut aussi provoquer l’activation de gènes, tels que les gènes spécifiques de la lignée germinale mâle, et de rétrotransposons normalement réprimés par la méthylation de l’ADN. Ceci entraîne la production d’antigènes habituellement absents des cellules somatiques et permet d’augmenter la visibilité de la tumeur par le système immunitaire. Le ciblage de ces antigènes cancer-spécifiques représente une autre voie thérapeutique qui est en cours de développement.
L’azacytidine nécessite d’être administrée à faible dose car des concentrations élevées de la molécule exercent une importante cytotoxicité, interfèrent avec la synthèse de l’ADN et provoquent des lésions à l’ADN [54]. Pour limiter ces effets indésirables, de nouveaux analogues de la cytidine sont développés. Ainsi, la zébularine est dépourvue de toxicité dans des modèles murins [55].
Par ailleurs, la réactivation des rétrotransposons, en particulier les rétrovirus endogènes (ERV), entraîne la formation d’ARN doubles brins cytoplasmiques qui sont détectés par les protéines de défense antivirale, aboutissant à l’apoptose de la cellule [56,57]. Actuellement, de nouvelles voies de développement thérapeutique utilisent la convergence des thérapies épigénétiques et des immunothérapies anticancéreuses par le biais du mimétisme viral, cet état cellulaire d’activation de la réponse antivirale déclenché par des acides nucléiques endogènes souvent dérivés de rétrotransposons transcrits de manière aberrante [58].
L’intelligence artificielle pour la détection non invasive des cancers
L’un des objectifs majeurs de la recherche contre le cancer est de réussir à discriminer un échantillon issu d’un patient atteint d’un cancer d’un échantillon sain, et, dans l’idéal, de prédire le tissu cancéreux. Dans cette optique, l’intelligence artificielle, avec notamment l’apprentissage automatique (en anglais, machine learning), connaît un essor important ces dernières années, en apportant des performances parfois comparables à celles d’experts, et avec l’avantage d’être automatisée et adaptable à de multiples formes de données.
Modèles d’apprentissage automatique et méthylome
L’apprentissage automatique peut se définir comme la capacité d’un algorithme à apprendre et à reconnaître des motifs à partir d’exemples représentatifs, et à utiliser efficacement ces informations pour prendre des décisions sur le statut de nouvelles données indépendantes [59]. Depuis l’article fondateur d’Alan Turing en 1950 [60], les algorithmes d’apprentissage automatique ont évolué, ouvrant la voie à de nouvelles perspectives pour le diagnostic et le suivi des cancers [61] (→).
(→) Voir le Repères de A. Benani, m/s n° 3, mars 2024, page 283
Cette évolution a été accélérée par l’émergence de bases de données donnant accès à de large jeux de données multi-omiques, telles que The Cancer Genome Atlas (TCGA). Ceci favorise l’utilisation de données multidimensionnelles dont les structures sont difficilement identifiables manuellement. De nombreux types de modèles d’apprentissage sont actuellement utilisés. On distingue les modèles d’apprentissage automatique supervisé, qui apprennent des structures à partir de données annotées, et les modèles d’apprentissage non supervisé, prenant en entrée des données non étiquetées. Dans le cadre de la détection des cancers, les échantillons sont annotés selon le statut de la maladie (sain vs issu d’un cancer), le type de cancer, le stade de la maladie, etc.
Des résultats de classification obtenus par différentes études utilisant méthylomes et modèles d’apprentissage automatique se sont révélés prometteurs. Ces approches détectent des profils de méthylation spécifiques aux cancers directement dans la fraction tumorale de l’ADN circulant (Tableau I). Par exemple, le test Galleri, qui cible environ 1 million de sites CpG et se fonde sur des modèles d’apprentissage supervisé, présente une sensibilité de détection moyenne de 55,2 % (pour plus de 50 types de cancers différents, tous stades confondus) et une spécificité de 99,3 %. Le tissu d’origine peut aussi être prédit, avec une précision de 93 % [50,51]. Les modèles d’apprentissage automatique peuvent également être utilisés pour sélectionner les marqueurs différentiellement méthylés [48,62,63] qui pourront ensuite être analysés ou intégrés dans un nouvel algorithme.
Approches non-invasives de détection des cancers couplant la méthylation différentielle et l’apprentissage automatique. Cette liste reprend les études citées dans le texte (voir également [44]). WGBS : séquençage au bisulfite du génome entier (whole genome bisulfite sequencing) ; DMR : région(s) différentiellement méthylée(s) (differentially methylated regions) ; cfMeDIP : immunoprécipitation d’ADN circulant méthylé (cell-free methylated DNA immunoprecipitation).
Résolutions des données de méthylome
La méthylation de l’ADN peut être analysée selon différentes résolutions. Pour une haute résolution, au niveau de la base nucléotidique, un processus de conversion de l’information épigénétique en une information génétique, chimique (BS-seq, pour bisulfite sequencing) ou enzymatique (EM-seq, pour enzymatic methyl-seq), est nécessaire lors de la préparation des librairies de séquençage. Cela permet de conserver l’information de méthylation de chaque site CpG au cours des amplifications. Couplé au séquençage haut débit, qui couvre chaque base, il est possible de construire des modèles prenant en entrée des données à l’échelle du CpG unique. Cette résolution permet d’identifier les sites les plus différentiellement méthylés [49]. Elle nécessite néanmoins une large couverture de séquençage, ce qui entraîne des coûts élevés, notamment pour des analyses à l’échelle globale du génome. Pour répondre à cette problématique, certaines études ont développé des stratégies ciblant des régions d’intérêts [45–47,52] ou regroupant des sites CpG se situant dans la même région et ayant un niveau de méthylation similaire [49,64]. Ceci permet d’obtenir une meilleure couverture de séquençage au niveau de ces clusters. D’autres études analysent les haplotypes de méthylation, définis comme des motifs de méthylation de sites CpG adjacents présents sur le même fragment d’ADN [37,52]. Les sites CpG adjacents dans les génomes de mammifères peuvent être co-méthylés en raison de l’activité processive des méthyltransférases. Ce statut de co-méthylation, inclus dans les haplotypes, permet de renforcer la détection du signal cancer-spécifique. En effet, des résultats de classification obtenus avec des haplotypes de méthylation se sont révélés meilleurs que ceux obtenus avec des niveaux de méthylation moyen à chaque site CpG. En particulier, l’étude de Guo et al. a démontré que les résultats fondés sur une méthylation moyenne par site CpG sont limités par un plus faible ratio signal/bruit [37].
Modèles d’apprentissage automatique mixtes et méthylome
Afin d’améliorer les performances de classification, plusieurs études ont porté sur des analyses multi-analytes17. Néanmoins, aucun modèle d’apprentissage automatique prenant en entrée une combinaison de marqueurs incluant des données épigénétiques afin de diagnostiquer un cancer à partir d’une biopsie liquide, n’a été développé. De tels modèles ont pourtant été établis pour d’autres données multiomiques, avec, par exemple, le test CancerSeek qui combine l’analyse de mutations et le taux de protéines sériques dans un même modèle afin de détecter avec une bonne précision huit types de cancers [65]. Les modèles actuels reposant sur l’utilisation d’épi-marqueurs juxtaposent plusieurs algorithmes qui sélectionnent les marqueurs différentiellement méthylés spécifiques aux cancers étudiés, puis procèdent à la classification des échantillons à partir de cette sélection [37,48,50,51,62]. Notre étude DIAMOND regroupe un modèle d’apprentissage prenant en entrée des variations de proportion d’haplotypes de méthylation, avec une analyse des altérations du nombre de copies (CNA), afin d’augmenter les performances du modèle [52]. Les résultats sont très prometteurs, notamment pour le cancer du sein, qui reste actuellement un des cancers les plus difficile à détecter par biopsies liquides.
Conclusion
La littérature décrit précisément les altérations des profils de méthylation de l’ADN retrouvées dans les cellules cancéreuses. Ces marques d’hyper- et d’hypo-méthylation ont de multiples origines qui sont documentées, mais dont la chronologie d’apparition reste difficile à appréhender. L’altération des profils de méthylation, qui apparaît dès les premières étapes de la carcinogenèse, permet potentiellement de détecter les cancers à des stades précoces. En effet, la méthylation de l’ADN est un biomarqueur qui permet de répondre aux défis que posent les approches ciblant uniquement la recherche de mutations, en particulier dans le cadre des méthodes non-invasives, qui nécessitent d’être extrêmement sensibles pour détecter de faibles charges d’ADN tumoral. La démocratisation des modèles d’apprentissage automatique fondés sur des données épigénétiques ouvrent la voie à des innovations qui, à terme, permettront le développement de tests diagnostiques plus universels et couvrant tous les types de cancers et tous les patients.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
MutL homolog 1
Breast cancer gene 1
O6-methylguanine-DNA methyltransferase
Adenomatous polyposis coli
Retinoblastoma 1
Von Hippel-Lindau tumor suppressor
Cyclin-dependent kinase inhibitor 2A
Gènes codant des facteurs de transcription de la famille des proto-oncogènes MYC
B-cell leukemia/lymphoma 2 like 10
Catenin beta 1
Insulin receptor substrate 2
Insulin like growth factor 2
Melanoma antigen gene family member C2
Synaptonemal complex protein 2
Séquences d’ADN hautement répétées, faisant partie de la famille des SINE (short interspersed elements)
ORF1p est une protéine de liaison à l’ARN de haute affinité qui forme une particule ribonucléoprotéique avec l’ARN L1.
Analyses portant sur différentes molécules présentes dans un même échantillon.
Références
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 2011 ; 11 : 726–34. [CrossRef] [PubMed] [Google Scholar]
- Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science 2017 ; 357 : eaal2380–10. [CrossRef] [PubMed] [Google Scholar]
- Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov 2022 ; 12 : 31–46. [CrossRef] [PubMed] [Google Scholar]
- Parreno V, Loubiere V, Schuettengruber B, et al. Transient loss of Polycomb components induces an epigenetic cancer fate. Nature 2024 ; 629 : 688–96. [CrossRef] [PubMed] [Google Scholar]
- Razin A, Riggs AD. DNA Methylation and Gene Function. Science 1980 ; 210 : 604–10. [CrossRef] [PubMed] [Google Scholar]
- Mattei AL, Bailly N, Meissner A. DNA methylation: a historical perspective. Trends Genet 2022 ; 38 : 676–707. [CrossRef] [PubMed] [Google Scholar]
- Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007 ; 447 : 425–32. [CrossRef] [PubMed] [Google Scholar]
- Weber M. Profils de méthylation de l’ADN dans les cellules normales et cancéreuses. Med Sci (Paris) 2008 ; 24 : 731–4. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Gene Dev 2002 ; 16 : 6–21. [CrossRef] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009 ; 462 : 315–22. [CrossRef] [Google Scholar]
- Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 2016 ; 30 : 733–50. [CrossRef] [PubMed] [Google Scholar]
- Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet 2018 ; 19 : 81–92. [CrossRef] [PubMed] [Google Scholar]
- Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet 2012 ; 13 : 679–92. [CrossRef] [PubMed] [Google Scholar]
- Berman BP, Weisenberger DJ, Aman JF, et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet 2012 ; 44 : 40–6. [CrossRef] [Google Scholar]
- Timp W, Bravo HC, McDonald OG, et al. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med 2014 ; 6 : 61. [CrossRef] [PubMed] [Google Scholar]
- Baylin SB, Jones PA. Epigenetic Determinants of Cancer. Cold Spring Harbor Perspectives in Biology 2016 ; 8 : a019505. [CrossRef] [PubMed] [Google Scholar]
- Saghafinia S, Mina M, Riggi N, et al. Pan-Cancer Landscape of Aberrant DNA Methylation across Human Tumors. Cell Rep 2018 ; 25 : 1066–80.e8. [CrossRef] [PubMed] [Google Scholar]
- Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet 2007 ; 16 : R50–9. [CrossRef] [PubMed] [Google Scholar]
- Sproul D, Kitchen RR, Nestor CE, et al. Tissue of origin determines cancer-associated CpG island promoter hypermethylation patterns. Genome Biol 2012 ; 13 : R84. [CrossRef] [PubMed] [Google Scholar]
- Gal-Yam EN, Egger G, Iniguez L, et al. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci USA 2008 ; 105 : 12979–84. [CrossRef] [PubMed] [Google Scholar]
- Stewart-Morgan KR, Requena CE, Flury V, et al. Quantifying propagation of DNA methylation and hydroxymethylation with iDEMS. Nat Cell Biol 2023 ; 25 : 183–93. [CrossRef] [PubMed] [Google Scholar]
- Zhou W, Dinh HQ, Ramjan Z, et al. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat Genet 2018 ; 50 : 591–602. [CrossRef] [PubMed] [Google Scholar]
- Court F, Boiteux EL, Fogli A, et al. Transcriptional alterations in glioma result primarily from DNA methylation-independent mechanisms. Genome Res 2019 ; 29 : 1605–21. [CrossRef] [PubMed] [Google Scholar]
- Widschwendter M, Fiegl H, Egle D, et al. Epigenetic stem cell signature in cancer. Nat Genet 2007 ; 39 : 157–8. [CrossRef] [PubMed] [Google Scholar]
- Carvalho DDD, Sharma S, You JS, et al. DNA Methylation Screening Identifies Driver Epigenetic Events of Cancer Cell Survival. Cancer Cell 2012 ; 21 : 655–67. [CrossRef] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell 2009 ; 136 : 823–37. [CrossRef] [PubMed] [Google Scholar]
- Rousseaux S, Reynoird N, Gaucher J, et al. L’intrusion des régulateurs de l’épigénome mâle dans les cellules somatiques cancéreuses. Med Sci (Paris) 2008 ; 24 : 735–41. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics 2009 ; 1 : 239–59. [CrossRef] [PubMed] [Google Scholar]
- Burns KH. Transposable elements in cancer. Nat Rev Cancer 2017 ; 17 : 415–24. [CrossRef] [PubMed] [Google Scholar]
- Rodic΄ N, Sharma R, Sharma R, et al. Long Interspersed Element-1 Protein Expression Is a Hallmark of Many Human Cancers. Am J Pathology 2014 ; 184 : 1280–6. [CrossRef] [Google Scholar]
- Lanciano S, Philippe C, Sarkar A, et al. Locus-level L1 DNA methylation profiling reveals the epigenetic and transcriptional interplay between L1s and their integration sites. Cell Genom 2024 ; 100498. [CrossRef] [PubMed] [Google Scholar]
- Taylor MS, Wu C, Fridy PC, et al. Ultrasensitive detection of circulating LINE-1 ORF1p as a specific multi-cancer biomarker. Cancer Discov 2023 ; 13 : 2532–47. [CrossRef] [PubMed] [Google Scholar]
- Rodriguez-Martin B, Alvarez EG, Baez-Ortega A, et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat Genet 2020 ; 52 : 306–19. [CrossRef] [PubMed] [Google Scholar]
- Scott EC, Gardner EJ, Masood A, et al. A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Res 2016 ; 26 : 745–55. [CrossRef] [PubMed] [Google Scholar]
- Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet 2006 ; 7 : 21–33. [CrossRef] [PubMed] [Google Scholar]
- Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer 2003 ; 3 : 253–66. [CrossRef] [PubMed] [Google Scholar]
- Guo S, Diep D, Plongthongkum N, et al. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat Genet 2017 ; 49 : 635–42. [CrossRef] [PubMed] [Google Scholar]
- Mouliere F. A hitchhiker’s guide to cell-free DNA biology. Neuro-Oncol Adv 2022 ; 4 : ii6–14. [CrossRef] [Google Scholar]
- Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014 ; 6 : 224ra24. [CrossRef] [PubMed] [Google Scholar]
- Heitzer E, Auinger L, Speicher MR. Cell-Free DNA and Apoptosis: How Dead Cells Inform About the Living. Trends Mol Med 2020 ; 26 : 519–28. [CrossRef] [PubMed] [Google Scholar]
- Snyder MW, Kircher M, Hill AJ, et al. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016 ; 164 : 57–68. [CrossRef] [PubMed] [Google Scholar]
- Moss J, Magenheim J, Neiman D, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun 2018 ; 9 : 5068. [CrossRef] [PubMed] [Google Scholar]
- Diaz LA, Bardelli A. Liquid Biopsies: Genotyping Circulating Tumor DNA. J Clin Oncol 2014 ; 32 : 579–86. [CrossRef] [PubMed] [Google Scholar]
- Luo H, Wei W, Ye Z, et al. Liquid Biopsy of Methylation Biomarkers in Cell-Free DNA. Trends Mol Med 2021 ; 27 : 482–500. [CrossRef] [PubMed] [Google Scholar]
- Shen SY, Singhania R, Fehringer G, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018 ; 563 : 579–83. [CrossRef] [PubMed] [Google Scholar]
- Nassiri F, Chakravarthy A, Feng S, et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat Med 2020 ; 26 : 1044–7. [CrossRef] [PubMed] [Google Scholar]
- Nuzzo PV, Berchuck JE, Korthauer K, et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat Med 2020 ; 26 : 1663. [CrossRef] [PubMed] [Google Scholar]
- Luo H, Zhao Q, Wei W, et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci Transl Med 2020 ; 12 : eaax7533. [CrossRef] [PubMed] [Google Scholar]
- Chen X, Gole J, Gore A, et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat Commun 2020 ; 11 : 3475. [CrossRef] [PubMed] [Google Scholar]
- Liu MC, Oxnard GR, Klein EA, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol 2020 ; 31 : 745–59. [CrossRef] [PubMed] [Google Scholar]
- Klein EA, Richards D, Cohn A, et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann Oncol 2021 ; 32 : 1167–77. [CrossRef] [PubMed] [Google Scholar]
- Michel, M. et al. Non-invasive multi-cancer detection using DNA hypomethylation of LINE-1 retrotransposons. Clin. Cancer Res. (2024) doi: 10.1158/1078-0432.ccr-24-2669. [Google Scholar]
- Jones PA, Issa J-PJ, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet 2016 ; 17 : 630–41. [CrossRef] [PubMed] [Google Scholar]
- Gnyszka A, Jastrzebski Z, Flis S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res 2013 ; 33 : 2989–96. [PubMed] [Google Scholar]
- Herranz M, Martín-Caballero J, Fraga MF, et al. The novel DNA methylation inhibitor zebularine is effective against the development of murine T-cell lymphoma. Blood 2006 ; 107 : 1174–7. [CrossRef] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015 ; 162 : 974–86. [CrossRef] [PubMed] [Google Scholar]
- Roulois D, Loo Yau H, Singhania R, et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015 ; 162 : 961–73. [CrossRef] [PubMed] [Google Scholar]
- Chen R, Ishak CA, Carvalho DDD. Endogenous Retroelements and the Viral Mimicry Response in Cancer Therapy and Cellular Homeostasis. Cancer Discov 2021 ; 11 : 2707–25. [CrossRef] [PubMed] [Google Scholar]
- Bhinder B, Gilvary C, Madhukar NS, et al. Artificial Intelligence in Cancer Research and Precision Medicine. Cancer Discov 2021 ; 11 : 900–15. [CrossRef] [PubMed] [Google Scholar]
- Turing AM. Computing machinery and intelligence. Mind 1950 ; LIX : 433–60. [CrossRef] [Google Scholar]
- Benani A. Histoire et perspectives de l’intelligence artificielle. Med Sci (Paris) 2024 ; 40 : 283–6. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Xu R, Wei W, Krawczyk M, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater 2017 ; 16 : 1155–61. [CrossRef] [PubMed] [Google Scholar]
- Liang W, Zhao Y, Huang W, et al. Non-invasive diagnosis of early-stage lung cancer using high-throughput targeted DNA methylation sequencing of circulating tumor DNA (ctDNA). Theranostics 2019 ; 9 : 2056–70. [CrossRef] [PubMed] [Google Scholar]
- Li W, Li Q, Kang S, et al. CancerDetector: ultrasensitive and non-invasive cancer detection at the resolution of individual reads using cell-free DNA methylation sequencing data. Nucleic Acids Res 2018 ; 46 : e89. [CrossRef] [PubMed] [Google Scholar]
- Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018 ; 1 : 3247–10. [Google Scholar]
Liste des tableaux
Approches non-invasives de détection des cancers couplant la méthylation différentielle et l’apprentissage automatique. Cette liste reprend les études citées dans le texte (voir également [44]). WGBS : séquençage au bisulfite du génome entier (whole genome bisulfite sequencing) ; DMR : région(s) différentiellement méthylée(s) (differentially methylated regions) ; cfMeDIP : immunoprécipitation d’ADN circulant méthylé (cell-free methylated DNA immunoprecipitation).
Liste des figures
|
Figure 1 Profils de méthylation différentiels caractéristiques des génomes tumoraux. A. Les génomes somatiques sains sont globalement méthylés, excepté au niveau des îlots CpG : la vaste majorité des îlots CpG n’est pas méthylée dans les génomes sains. Ceci concerne également 72 % des promoteurs associés à un îlots CpG, quel que soit le statut d’expression du gène régulé par ce promoteur [6]. B. Les génomes tumoraux combinent hypométhylation et hyperméthylation acquises au cours de la carcinogenèse (illustration conçue avec BioRender.com). |
| Dans le texte | |
|
Figure 2 Défis et innovations en matière de détection des altérations tumorales à visée diagnostique : le potentiel de la méthylation de l’ADN circulant. Les progrès réalisés dans le domaine de la biopsie liquide ont démontré l’intérêt de quantifier l’ADN circulant tumoral (ADNct) pour améliorer la prise en charge des patients. Cependant, détecter les faibles fractions d’ADN tumoral, libérées aux stades précoces ou au cours du traitement, reste un défi et nécessite le développement de méthodes de détection et de quantification plus sensibles (illustration conçue avec BioRender.com). |
| Dans le texte | |
|
Figure 3 Biopsies liquides et ADN tumoral circulant (ADNct). L’ADN circulant (ADNc) est présent dans le système sanguin sous forme de fragments libres, qui peuvent être isolés à partir du plasma (la phase acellulaire du sang). Ces fragments présentent une taille moyenne de 167 paires de bases (bp), équivalente à la taille d’un brin d’ADN enroulé autour d’un nucléosome, ce qui suggère qu’ils sont majoritairement générés au cours de la dégradation du génome lors de la mort cellulaire par apoptose. Chez les patients atteints de cancer, une fraction de cet ADN plasmatique provient de la tumeur (illustration conçue avec BioRender.com). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.