")
")
| Issue |
Med Sci (Paris)
Volume 40, Number 10, Octobre 2024
|
|
|---|---|---|
| Page(s) | 707 - 710 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2024110 | |
| Published online | 25 October 2024 | |
CAPS ou pas CAPS ?
La diversité fonctionnelle des mutations gain-de-fonction de NLRP3 modifie l’approche diagnostique des auto-inflammations héréditaires CAPS
Functional diversity of NLRP3 gain-of-function mutations and impact on the diagnosis of CAPS hereditary auto-inflammation disease
Centre international de recherche en infectiologie (CIRI), Inserm U1111, Université Claude Bernard Lyon 1, CNRS UMR5308, École normale supérieure de Lyon Lyon France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Les enjeux du diagnostic des maladies auto-inflammatoires associées à NLRP3
Trois maladies auto-inflammatoires associées à NLRP3 (anciennement nommées syndromes périodiques associés à la cryopyrine, CAPS) ont été décrites : 1) l’autoinflammation familiale au froid, la plus bégnine des trois, caractérisée par des manifestations cutanées et l’arthralgie ; 2) le syndrome de Muckle-Wells, caractérisé par l’ajout d’une atteinte des organes sensoriels (surdité, inflammation oculaire), d’une arthrite, et de migraines ; et 3) la maladie inflammatoire multisystémique à début néonatal (également nommée syndrome chronique infantile neurologique, cutané et articulaire), qui est caractérisée par une inflammation du système nerveux central avec des méningites chroniques et une inflammation des articulations causant des arthropathies déformantes, en plus des autres manifestations déjà citées [1]. En réalité, les frontières nosologiques entre ces trois maladies sont assez mal définies car les patients présentent des combinaisons variées des différents symptômes (Figure 1). L’amylose AA inflammatoire est une complication commune à ces maladies.
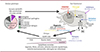 |
Figure 1. Nouveau test fonctionnel pour les variants de NLRP3 : applications pour le diagnostic et le traitement des maladies auto-inflammatoires associées à ces variants. Le test de l’activité de l’inflammasome (mort cellulaire et sécrétion de IL(interleukine)-1β et IL-18 après l’expression de variants de NLRP3 dans la lignée monocytaire U937, avec et sans amorçage (par du lipopolysaccharide bactérien, LPS) ou signal d’activation (nigéricine) de la protéine, permet de caractériser les différents gains de fonction des mutants pathogènes, ce qui fournit des informations indispensables pour le diagnostic des maladies auto-inflammatoires associées à ces variants (NLRP3-associated autoinflammatory diseases, NLPR3-AID), et pour un accès argumenté des patients au traitement par des antagonistes de l’interleukine-1 (anti-IL-1). De plus, ce test standardisé permet le criblage d’inhibiteurs pharmacologiques de NLRP3 sur les différents variants, et contribue ainsi à prédire la réponse des patients aux futures molécules thérapeutiques. FCAS : autoinflammation familiale au froid ; NOMID : maladie inflammatoire multisystémique à début néonatal ; VUS : variant de signification incertaine. |
Ces maladies sont causées par des mutations gain-de-fonction de NLRP3, codant un senseur de stress cellulaire qui assemble l’inflammasome [2] (→).
(→) Voir la Synthèse de M. Groslambert et B.F. Py, m/s n° 1, janvier 2018, page 47
Ce complexe de signalisation contrôle la sécrétion des cytokines inflammatoires IL(interleukine)-1β et IL-18, ainsi que la mort cellulaire par pyroptose, libérant de nombreuses alarmines inflammatoires. Ces mutations sont le plus souvent héritées, mais elles peuvent aussi se produire de novo dans les cellules somatiques de l’embryon, notamment dans le cas de la maladie inflammatoire multisystémique à début néonatal. Elles entraînent une dérégulation de la sécrétion d’IL-1β, qui rend compte de la plupart des symptômes, comme en témoigne l’efficacité du traitement par un antagoniste de l’IL-1 chez la majorité des patients. Cependant, ce traitement est inefficace contre les lésions déjà constituées, et la prise en charge des patients devrait donc bénéficier d’un diagnostic plus précoce. Le diagnostic clinique repose sur une augmentation des niveaux de protéines de la phase aiguë de l’inflammation (en anglais, acute phase proteins) et sur la présence d’au moins deux des critères suivants : urticaire, déclenchement au froid, surdité neurosensorielle, méningite non infectieuse, symptômes musculo-squelettiques. Le résultat de l’analyse génétique moléculaire corrobore le diagnostic clinique. Les variants de NLRP3 sont classés comme pathogéniques, probablement pathogéniques, bénins, probablement bénins, ou comme variants de signification incertaine, d’après leur fréquence dans la population, leur association récurrente avec la maladie, la ségrégation familiale, ainsi que des prédictions de pathogénicité fondées sur des algorithmes in silico et des données fonctionnelles [3] (Figure 1). Néanmoins, la pathogénicité reste débattue pour la majorité des variants recensés dans les bases de données Infevers1 et ClinVar2 [4]. Pour cette maladie rare (1/360 000 naissances en France), à pénétrance incomplète, l’identification des mutations gain-de-fonction nécessite des tests fonctionnels, qui reposent traditionnellement sur la mesure de la sécrétion d’IL-1β, en réponse au lipopolysaccharide bactérien, par des cellules exprimant le variant de NLRP3 contenant la mutation. Le lipopolysaccharide est utilisé comme activateur du facteur de transcription NF-kB (nuclear factor-kappa B) pour induire l’expression de la pro-IL-1β. Néanmoins, il permet aussi « l’amorçage » de la protéine NLRP3, qui augmente sa synthèse et la sensibilise, par des modifications post-traductionnelles, à différents signaux d’activation, que l’on peut classer selon le stress cellulaire qu’ils induisent [2]. Ces divers stress cellulaires contrôlent l’assemblage de l’inflammasome. Ainsi, ces tests ne permettent pas de distinguer les gains de fonction de NLRP3 correspondant à une sensibilisation de la protéine aux signaux d’activation en l’absence de signal d’amorçage, ni de distinguer les mutants à activité constitutive ou déclenchée par l’amorçage sans signal d’activation.
Hétérogénéité des mutants gainde-fonction de NLRP3 quant à leur dépendance aux signaux d’amorçage et d’activation
Pour caractériser les variants de NLRP3 fonctionnellement, nous avons développé un test cellulaire standardisé dans lequel l’expression, l’amorçage et l’activation de NLRP3 sont contrôlés par des signaux distincts [5]. Des cellules de la lignée monocytaire humaine U937, invalidées pour NLRP3, ont été complémentées par des variants naturels de ce gène inductibles par la doxycycline. Nous avons ensuite mesuré l’activité de la protéine variante en réponse à l’induction du gène par la doxycycline, en l’absence ou en présence d’un signal d’amorçage fourni par le lipopolysaccharide, ou d’un signal d’activation fourni par la nigéricine (une toxine bactérienne activatrice de NLRP3) (Figure 1). Avec ce test standardisé, nous avons réalisé la plus grande étude fonctionnelle comparative des variants de NLRP3 (34 variants différents) à ce jour, et les principaux résultats ont été validés sur des monocytes de patients (représentant 11 variants). Par groupement non biaisé, les variants sans retentissement fonctionnel ont été différenciés des mutations gain-de-fonction pathogéniques. Celles-ci se sont révélées hétérogènes. Alors que certains mutants de NLRP3 présentent une activité constitutive dès leur expression, l’activité des autres mutants nécessite un stimulus supplémentaire : signal d’amorçage ou d’activation indifféremment, signal d’amorçage spécifiquement, ou signal d’activation spécifiquement [5]. Ces derniers mutants, activés par un signal d’activation indépendamment de tout amorçage, n’étaient pas identifiés par les tests précédents [6]. Certains de ces mutants sont d’ailleurs relativement fréquents dans la population générale (par exemple, le mutant NLRP3Q703K [glutamine en position 703 substituée par une lysine] est présent chez environ 1 personne pour 2000 (0,04 %), mais ils ne sont pas toujours associés à la maladie, et correspondraient donc plutôt à des allèles hypermorphes3 de susceptibilité à l’autoinflammation. Ils sont parfois associés à des manifestations cliniques atypiques, par leur apparition à l’âge adulte, ou par leur nature même (e.g., surdité en l’absence d’atteinte cutanée ou de fièvre) [6]. Ces nouvelles données invitent donc à revoir le diagnostic et les options thérapeutiques pour ces personnes. Le lien entre le profil d’activation des mutants de NLRP3 dans un test cellulaire et les manifestations cliniques de la mutation est assez lâche. En effet, même si les manifestations cliniques sont en partie corrélées avec le génotype, elles peuvent varier fortement au sein d’une même famille. De plus, depuis l’utilisation thérapeutique des antagonistes d’IL-1, la sévérité des symptômes est plus fortement liée au délai de prise en charge des personnes atteintes de la maladie qu’à la nature de la mutation. Néanmoins, la plupart (4 sur 5) des mutations « somatiques »4 de NLRP3 qui étaient accompagnées de symptômes correspondaient à des protéines constitutivement actives [5]. Ces mutations ne sont qu’exceptionnellement héritées et présentes dans toutes les cellules de l’individu, probablement parce qu’alors incompatibles avec sa survie.
Hétérogénéité des mutants de NLRP3 quant à leur sensibilité aux inhibiteurs pharmacologiques
Plusieurs inhibiteurs directs de NLRP3 sont en cours de développement, mais la sensibilité des mutants pathogéniques de NLRP3 au composé phare MCC950 est débattue [7, 8]. Par le même test que celui utilisé précédemment pour la caractérisation fonctionnelle des mutations, nous avons mesuré la sensibilité de chacun des 34 variants de NLRP3 à deux inhibiteurs directs, MCC950 et CY-09, et deux inhibiteurs indirects, G5 et CRT0066101, ciblant des modifications post-traductionnelles de la protéine [9, 10]. Bien que minoritaires, quelques mutants étaient résistants à MCC950 (5 sur 34), qui cible une région impliquée dans le changement conformationnel accompagnant l’activation de NLRP3, une région qui concentre les mutations. En revanche, tous les mutants étaient sensibles à CY-09, un inhibiteur dont pourraient donc bénéficier un plus grand nombre de patients. Enfin, en absence d’amorçage, tous les mutants étaient résistants à CRT0066101, ce qui suggère l’existence, dans ces conditions, d’une voie d’activation alternative de NLRP3, en accord avec une étude récente décrivant deux voies distinctes d’assemblage de l’inflammasome [5, 11].
La caractérisation des mutants gain-de-fonction de NLRP3 permet d’identifier les domaines clés pour le contrôle de l’assemblage de l’inflammasome
L’inflammasome NLRP3 est un acteur important de l’inflammation délétère associée à de nombreuses maladies communes, métaboliques (diabète de type 2), cardiovasculaires (athérosclérose) ou neurodégénératives (maladie d’Alzheimer) [12]. Une meilleure compréhension de son mécanisme d’activation est un prérequis pour le développement d’inhibiteurs spécifiques à fort potentiel thérapeutique. La plupart des études mécanistiques reposent sur des modèles murins, et la caractérisation fonctionnelle des mutations responsables des maladies auto-inflammatoires associées à NLRP3 apporte des données complémentaires chez l’homme. L’activation physiologique de NLRP3 résulte d’un échange ADP/ATP conduisant à un changement conformationnel majeur de la protéine. Comme attendu, les mutations gain-de-fonction de NLRP3 sont concentrées au site ATPase de la protéine, et aux interfaces entre sous-domaines impliqués dans ce changement conformationnel. De plus, nous avons identifié un sillon à la surface de la protéine (donc a priori accessible à d’éventuelles molécules à visée thérapeutique), comprenant six acides aminés consécutifs, dont les mutations conduisent à une activité constitutive de NLRP3 [5]. Nous tenterons d’élucider son rôle dans le contrôle de l’assemblage de l’inflammasome, afin de saisir l’opportunité de le cibler pharmacologiquement. Enfin, l’accumulation des mutations gain-de-fonction dans le domaine « leucin-rich repeats (LRR) » C-terminal de NLRP3 suggère que le maintien d’une certaine boucle acide dans la phase concave du LRR, stabilise la forme inactive de la protéine [13]. Ainsi, notre analyse des variants naturels de la protéine NLRP3 humaine fournit de nouvelles informations sur les différents domaines contrôlant son activation (Figure 2).
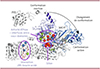 |
Figure 2. Identification des structures impliquées dans l’activation de NLRP3 par l’analyse de ses variants naturels. Les mutations gain-de-fonction de NLRP3 sont concentrées au site ATPase de la protéine, aux interfaces entre sous-domaines impliqués dans le changement conformationnel responsable de son activation, dans un sillon accessible à la surface de la protéine, sans fonction connue, et à l’interface entre le domaine leucine-rich repeats (LRR) et une boucle acide (en rouge). La mise en évidence de ces structures clés pour le contrôle de l’activation de NLRP3 permettra d’orienter les recherches mécanistiques et de les cibler pharmacologiquement. Les sphères colorées indiquent les mutations gain-de-fonction de NLRP3 responsables d’une activité constitutive de la protéine (en rouge), d’une activité dépendante indifféremment de l’amorçage ou du signal d’activation (en vert), d’une activité dépendante strictement de l’amorçage (en jaune) ou strictement du signal d’activation (en bleu), ainsi que les variants sans gain de fonction (en gris). PYD : domaine pyrine ; NACHT : domaine nommé d’après les autres protéines qui le possèdent (NAIP, CIITA, HET-E et TEP1). |
Perspectives
La caractérisation fonctionnelle des variants naturels de NLRP3 est actuellement indispensable au diagnostic des maladies auto-inflammatoires qui leur sont associées. L’analyse fonctionnelle d’un plus grand nombre de mutants, l’affinement de notre compréhension des mécanismes structuraux de l’activation de cette protéine, et les progrès de l’analyse par l’intelligence artificielle, pourraient permettre de prédire in silico la pathogénicité de nouveaux variants de NLRP3, comme le propose l’algorithme d’apprentissage automatique Alpha-Missense [14] (→).
(→) Voir la Chronique génomique de B. Jordan, m/s n° 12, décembre 2023, page 981
Les outils que nous avons développés sont directement applicables pour le développement de nouveaux inhibiteurs pharmacologiques de NLRP3, en utilisant le profil de résistance des différents variants de la protéine à ces inhibiteurs comme signature de leurs mécanismes d’action et des régions ciblées. Dans le futur, cette approche pourrait également permettre de prédire, pour chaque mutation, l’efficacité de différentes thérapies.
Liens d’intérêt
Les auteurs déclarent un brevet EP23306463.3 (Methods for the diagnosis, treatment and analysis of the NLRP3-associated autoinflammatory diseases) déposé par Inserm-Transfert (BP).
On qualifie d’hypermorphe un allèle qui correspond à une surexpression du gène sauvage ou qui code une forme hyperactive de son produit
Ce terme désigne les mutations survenant dans une cellule non germinale. À la différence des mutations survenant dans une cellule de la lignée germinale, les mutations somatiques ne peuvent pas être transmises à la descendance.
Références
- Booshehri LM, Hoffman HM. CAPS and NLRP3. J Clin Immunol 2019 ; 39 : 277–86. [CrossRef] [PubMed] [Google Scholar]
- Groslambert M, Py BF. NLRP3, un inflammasome sous contrôle. Med Sci (Paris) 2018 ; 34 : 47–53. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Van Gijn ME, Ceccherini I, Shinar Y, et al. New workflow for classification of genetic variants’ pathogenicity applied to hereditary recurrent fevers by the International Study Group for Systemic Autoinflammatory Diseases (INSAID). J Med Genet 2018 ; 55 : 530–7. [CrossRef] [PubMed] [Google Scholar]
- Milhavet F, Cuisset L, Hoffman HM, et al. The infevers autoinflammatory mutation online registry: update with new genes and functions. Hum Mutat 2008 ; 29 : 803–8. [CrossRef] [PubMed] [Google Scholar]
- Cosson C, Riou R, Patoli D, et al. Functional diversity of NLRP3 gain-of-function mutants associated with CAPS autoinflammation. J Exp Med 2024 ; 221 : e20231200. [CrossRef] [PubMed] [Google Scholar]
- Fayand A, Cescato M, Le Corre L, et al. Pathogenic variants in the NLRP3 LRR domain at position 861 are responsible for a boost-dependent atypical CAPS phenotype. J Allergy Clin Immunol 2023 ; 152 : 1303–11.e1. [CrossRef] [PubMed] [Google Scholar]
- Vande Walle L, Stowe IB, Šácha P, et al. MCC950/ CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease-associated mutants for inflammasome inhibition. PLoS Biol 2019 ; 17 : e3000354. [CrossRef] [PubMed] [Google Scholar]
- Weber ANR, Tapia-Abellán A, Liu X, et al. Effective ex vivo inhibition of cryopyrin-associated periodic syndrome (CAPS)-associated mutant NLRP3 inflammasome by MCC950/CRID3. Rheumatol Oxf Engl 2022 ; 61 : e299–e313. [CrossRef] [PubMed] [Google Scholar]
- Py BF, Kim MS, Vakifahmetoglu-Norberg H, et al. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 2013 ; 49 : 331–8. [CrossRef] [PubMed] [Google Scholar]
- Zhang Z, Meszaros G, He WT, et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J Exp Med 2017 ; 214 : 2671–93. [CrossRef] [PubMed] [Google Scholar]
- Mateo-Tórtola M, Hochheiser IV, Grga J, et al. Non-decameric NLRP3 forms an MTOC-independent inflammasome. BioRxiv 2023 ; 2023.07.07.548075. [Google Scholar]
- Mangan MSJ, Olhava EJ, Roush WR, et al. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov 2018 ; 17 : 588–606. [CrossRef] [PubMed] [Google Scholar]
- Hochheiser IV, Pilsl M, Hagelueken G, et al. Structure of the NLRP3 decamer bound to the cytokine release inhibitor CRID3. Nature 2022 ; 604 : 184–9. [CrossRef] [PubMed] [Google Scholar]
- Jordan B. Les secrets des variants. Med Sci (Paris) 2023 ; 39 : 981–3. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
© 2024 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Liste des figures
|
Figure 1. Nouveau test fonctionnel pour les variants de NLRP3 : applications pour le diagnostic et le traitement des maladies auto-inflammatoires associées à ces variants. Le test de l’activité de l’inflammasome (mort cellulaire et sécrétion de IL(interleukine)-1β et IL-18 après l’expression de variants de NLRP3 dans la lignée monocytaire U937, avec et sans amorçage (par du lipopolysaccharide bactérien, LPS) ou signal d’activation (nigéricine) de la protéine, permet de caractériser les différents gains de fonction des mutants pathogènes, ce qui fournit des informations indispensables pour le diagnostic des maladies auto-inflammatoires associées à ces variants (NLRP3-associated autoinflammatory diseases, NLPR3-AID), et pour un accès argumenté des patients au traitement par des antagonistes de l’interleukine-1 (anti-IL-1). De plus, ce test standardisé permet le criblage d’inhibiteurs pharmacologiques de NLRP3 sur les différents variants, et contribue ainsi à prédire la réponse des patients aux futures molécules thérapeutiques. FCAS : autoinflammation familiale au froid ; NOMID : maladie inflammatoire multisystémique à début néonatal ; VUS : variant de signification incertaine. |
| Dans le texte | |
|
Figure 2. Identification des structures impliquées dans l’activation de NLRP3 par l’analyse de ses variants naturels. Les mutations gain-de-fonction de NLRP3 sont concentrées au site ATPase de la protéine, aux interfaces entre sous-domaines impliqués dans le changement conformationnel responsable de son activation, dans un sillon accessible à la surface de la protéine, sans fonction connue, et à l’interface entre le domaine leucine-rich repeats (LRR) et une boucle acide (en rouge). La mise en évidence de ces structures clés pour le contrôle de l’activation de NLRP3 permettra d’orienter les recherches mécanistiques et de les cibler pharmacologiquement. Les sphères colorées indiquent les mutations gain-de-fonction de NLRP3 responsables d’une activité constitutive de la protéine (en rouge), d’une activité dépendante indifféremment de l’amorçage ou du signal d’activation (en vert), d’une activité dépendante strictement de l’amorçage (en jaune) ou strictement du signal d’activation (en bleu), ainsi que les variants sans gain de fonction (en gris). PYD : domaine pyrine ; NACHT : domaine nommé d’après les autres protéines qui le possèdent (NAIP, CIITA, HET-E et TEP1). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.