")
")
| Issue |
Med Sci (Paris)
Volume 36, Number 11, Novembre 2020
|
|
|---|---|---|
| Page(s) | 1012 - 1017 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2020173 | |
| Published online | 05 November 2020 | |
Les myélinosomes : une nouvelle voie du contrôle de qualité des protéines
Myelinosomes: A new pathway of protein quality control
1 Sechenov Institut of Evolutionary Physiology and Biochemistry, Russian Academy of Sciences, 44 Thoreza pr., 194223, St-Petersburg, Russie
2 CHU de Rennes, Département de gynécologie obstétrique et reproduction humaine-CECOS, Hôpital Sud, 16 boulevard de Bulgarie, 35000 Rennes, France
3 Univ Rennes, Inserm, EHESP, Irset (Institut de recherche en santé, environnement et travail) -UMR_S 1085, F-35000 Rennes, France
4 Univ Poitiers, Plateforme ImageUP, 1 rue Georges Bonnet, F-86022 Poitiers, France
5 Laboratoire STIM, Équipe 4CS, ERL CNRS 7003. 1 rue Georges Bonnet 86022 Poitiers Cedex, France
6 CHU de Poitiers, Poitiers F-86021, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Deux voies de dégradation des protéines mal repliées sont classiquement décrites : la voie du protéasome et la voie de l’autophagie. Nous décrivons ici une nouvelle voie de protéostase cellulaire ne dégradant pas la protéine anormale mais l’expulsant hors de la cellule grâce à des nanovésicules appelées myélinosomes. Ces myélinosomes sont produits par la cellule dans des situations pathologiques ou de stress en lien avec des facteurs génétiques ou environnementaux. Sur le plan morphologique, les myélinosomes sont caractérisés par des membranes osmiophiles denses aux électrons dont l’arrangement empilé est semblable à celui de la myéline et présente jusqu’à 30 feuillets selon le type de cellule. Dans deux modèles, au moins, de maladies génétiques (la maladie de Huntington et la mucoviscidose), les myélinosomes sont importants pour éliminer les protéines mutées par un processus sécrétoire inhabituel, évitant ainsi leur agrégation dans les cellules.
Abstract
Maintenance of cell proteostasis relies on two degradation pathways: proteasome and autophagy. Here we describe a new proteostasis pathway avoiding degradation of abnormal proteins yet carrying them outside the cell using nanovesicles called myelinosomes. These myelinosomes are produced in pathological or stress situations in relation with genetic or environmental factors. Myelinosome vesicles are nano-sized multi-stacked membrane structures, resembling myelin sheath. It has recently been shown in two models of genetic diseases (Huntington’s disease and cystic fibrosis) that myelinosomes are important for eliminating mutant proteins in an unusual secretory process, thus preventing their accumulation and aggregation in cells.
© 2020 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (Photo © Nicolas Bourmeyster).
Après leur synthèse par les ribosomes, les protéines subissent un processus de repliement leur permettant d’acquérir leur structure fonctionnelle. Ce repliement, généralement spontané dans les conditions physiologiques, se produit à un rythme qui dépend de la taille de la protéine. Les premières étapes du processus comprennent le regroupement d’acides aminés hydrophobes et l’expulsion de molécules d’eau, puis le compactage de la chaîne polypeptidique et la consolidation de la structure secondaire et enfin une réorganisation qui donne à la protéine sa structure tertiaire finale. De nombreux facteurs et co-facteurs, comme les protéines chaperonnes, les cations métalliques ou certaines modifications post-traductionelles, influent sur l’issue du processus de repliement [1] (→).
(→) Voir la Synthèse de A.P. Arrigo, m/s n° 6-7, juin-juillet 2005, page 619
Un mauvais repliement des protéines est un événement fréquent dans les cellules normales ; les protéines mal repliées sont alors éliminées par des systèmes de contrôle de qualité des protéines (CQP). Lorsque la production de protéines dépasse la capacité de contrôle de qualité, ce qui survient dans les cellules âgées ou atteintes de certaines altérations génétiques, une surcharge apparaît avec l’accumulation dans la cellule de protéines mal repliées. En fonction de ses propriétés et de l’efficacité des systèmes CQP, la protéine qui s’est accumulée peut être soit dégradée, soit assemblée en oligomères et agrégats toxiques. Au cours de cette agrégation, les protéines mal repliées peuvent recruter et inactiver d’autres protéines fonctionnelles, entraînant alors des dommages cellulaires irréversibles.
La dégradation des protéines peut être réalisée dans le protéasome, un complexe composé de plus de trente protéines différentes. La destruction de la protéine mal repliée a alors lieu dans le canal du protéasome dans lequel elle est découpée en peptides de 2 ou 3 acides aminés [2] (→).
(→) Voir l’Éditorial de N. Benaroudj, m/s n° 1, janvier 2005, page 115
Les protéines destinées à être ainsi dégradées sont identifiées par l’ajout sur leur séquence d’une chaîne d’ubiquitine. Ce petit polypeptide, polymérisé au niveau d’une lysine de la protéine aberrante, permet alors sa reconnaissance par le protéasome. Les dysfonctionnements de ce système protéasome-ubiquitine sont généralement associés à un stress protéotoxique1. La diminution de son efficacité, au cours du vieillissement, semble ainsi jouer un rôle-clé dans la survenue de maladies neurodégénératives dépendantes de l’âge.
Un autre mode de dégradation et de recyclage permettant d’assurer la protéostase repose sur l’activation de l’autophagie [3] (→) littéralement « se manger soi-même », l’autophagie permet la dégradation des protéines en acides aminés qui seront ensuite recyclés dans le cytoplasme de la cellule. Une vésicule, l’autophagosome, formée à partir d’un fragment de membrane interne, le phagophore, s’allonge et se referme sur les constituants cytoplasmiques destinés à être dégradés. Cette vacuole à double membrane fusionne ensuite avec des lysosomes qui acidifient son contenu et le dégradent. Les produits obtenus sont finalement libérés dans le cytoplasme pour être réutilisés par la cellule. L’autophagosome est une structure caractéristique d’environ 0,5 à 1 μm de diamètre qui présente un contenu hétérogène. Si la formation du phagophore est initiée au niveau du réticulum endoplasmique, d’autres membranes cellulaires, notamment des membranes mitochondriales [4] (→), sont impliquées dans sa constitution. Un type particulier d’autophagie, l’autophagie relayée par des protéines chaperonnes (CMA), dégrade sélectivement des protéines présentant une séquence précise de 5 acides aminés (KFERQ). Ces protéines, prises en charge par un complexe formé par les protéines chaperonnes (hsc70) et co-chaperonnes (hip, hop, hsp40, hsp90 et bag1), sont dans ce cas dirigées vers le lysosome où LAMP-2A, un récepteur associé à sa membrane, les reconnaît, permettant leur translocation dans la lumière lysosomale [3]. Dans les cellules présentatrices d’antigènes, un autre mode de dégradation dépendant des lysosomes implique les molécules du complexe majeur d’histocompatibilité de classe II (CMHII). Ce mécanisme assure la dégradation de protéines d’origines intracellulaire ou extracellulaire (ingérées par exemple lors de la phagocytose) dans des compartiments apparentés aux lysosomes, ce qui conduit à la présentation des peptides obtenus à la surface des cellules [5].
Toute résistance à la dégradation par les mécanismes de protéolyse cellulaire, dépendants (protéolyse lysosomale induite par autophagie ou via le CMHII) ou indépendants (système protéasome-ubiquitine) du lysosome, entraîne une accumulation d’agrégats protéiques, perturbe la protéostase cellulaire, et interfère avec le métabolisme cellulaire normal.
D’autres mécanismes de contrôle qualité commencent à être identifiés, reflet de la complexité et de la dynamique du protéome des cellules eucaryotes. Des voies non conventionnelles de sécrétion de protéines normales et anormales ont en effet été décrites [6, 7]. Nous présentons ici une nouvelle voie de protéostase cellulaire. Contrairement aux autres voies, celle-ci ne dégrade pas la protéine anormale mais l’expulse intacte hors de la cellule, par l’intermédiaire de vésicules particulières, les myélinosomes. Il s’agit d’une nouvelle stratégie de maintien de la protéostase cellulaire sans dégradation protéique, contrairement aux stratégies classiques faisant intervenir le protéasome ou l’autophagie, qui reposent sur la dégradation des protéines mutées présentant une anomalie de conformation. Au cours de ce nouveau processus, les protéines mutées/mal repliées et non dégradées quittent la cellule intactes, véhiculées par les myélinosomes (Figure 1).
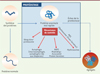 |
Figure 1. Différents mécanismes de contrôle de la qualité des protéines. Les protéines mal repliées sont repérées par différents mécanismes de contrôle comme le système ubiquitine/protéasome, l’autophagie (macroautophagie ou autophagie liée aux chaperonnes) ou le mécanisme MAPS (misfolded-associated protein secretion). Un nouveau mécanisme consiste en l’incorporation des protéines mal repliées au sein de myélinosomes qui sont ensuite éliminés par sécrétion extracellulaire (MDS). En cas d’échec ou de débordement de ces mécanismes de contrôle, les protéines anormales s’accumulent et forment des agrégats toxiques. |
Identification des myélinosomes
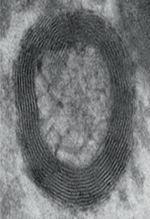
La première observation de ces organites date de 1989 [8]. Le terme « myélinosome » qui leur a été donné évoque l’aspect caractéristique en microscopie électronique des membranes empaquetées osmiophiles2 nettement denses aux électrons qui les composent. Les myélinosomes se présentent comme des vésicules délimitées par une paroi de structure lamellaire régulièrement ordonnée, constituée par l’alternance de lignes denses et claires évocatrices de la myéline, même si elles n’en comprennent pas (Figure 2). Les myélinosomes sont généralement constitués de deux zones : une couche externe formée d’un réseau de membranes concentriques, et une zone interne lucide aux électrons partiellement cloisonnée par des structures non-osmiophiles. Les citernes du réticulum endoplasmique rugueux (RER) et probablement la membrane nucléaire sont directement associées à l’apparition et au développement des myélinosomes.
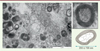 |
Figure 2. Ultrastructure des myélinosomes. Photographie en microscopie électronique de milieu de culture de cellules de Sertoli. On constate au-delà des myélinosomes (voir insert) la présence de nombreuses autres vésicules extracellulaires (microvésicules et exosomes). Insert : à fort grossissement, on constate la présence de nombreuses membranes superposées entourant une matrice contenant une structure lucide aux électrons ainsi qu’une forme de cloisonnement. |
La production de myélinosomes a été reliée, dès 1993, à une souffrance cellulaire avec la mise en évidence de leur formation en lien direct avec le RER et l’enveloppe nucléaire, par des hépatocytes en culture exposés à la quinacrine, un médicament antipaludéen amphiphile [9]. La quinacrine agissant sur le métabolisme des phospholipides, les auteurs conclurent alors que cette production abondante de myélinosomes était la réponse de la cellule à une agression du métabolisme des phospholipides. La quinacrine interagit en effet directement avec les phospholipides constitutifs du réticulum endoplasmique, de la membrane plasmique et des lysosomes. Ainsi altérés, les phospholipides s’accumulent et sont reconnus par la cellule comme anormaux. Ils se concentrent alors dans des vésicules semblables aux myélinosomes [9] qui avaient été décrits [8]. Rares dans les cellules normales, les myélinosomes intracellulaires apparaissent dans des situations pathologiques causées par des facteurs génétiques ou environnementaux. Ils sont d’ailleurs détectés en abondance dans les maladies dites d’agrégation. Un grand nombre de ces membranes osmiophiles denses aux électrons se retrouve également dans les maladies de surcharge lysosomale [10]. Ils pourraient constituer un nouvel organite de « stockage », à côté des autolysosomes, des endolysosomes et des endosomes tardifs [10].
Les myélinosomes, des organites sécrétoires
Les myélinosomes sont des organites sécrétés capables de quitter la cellule dans leur intégralité, sous forme de vésicules extracellulaires. Ils se différencient ainsi des autres organites sécrétoires, notamment ceux apparentés aux lysosomes (lysosome-related-organelles, LRO), comme les granules de sécrétion des cellules immunitaires du sang, des neurones, des cellules neuroendocrines, les corps lamellaires des kératinocytes pulmonaires ou cutanés, les mélanosomes ou encore les lysosomes sécrétoires [11]. Tous ces organites libèrent leur contenu dans l’espace extracellulaire après leur fusion avec la membrane plasmique, mais aucun n’est retrouvé sous forme d’organites intacts hors de la cellule, comme le sont les myélinosomes.
Les myélinosomes se forment à partir du RER [9] et leur trafic intracellulaire s’apparente à celui des corps lamellaires présents dans les cellules épithéliales pulmonaires qui se développent et se propagent dans les corps multivésiculaires (multi-vesicular bodies, MVB) [12]. Au niveau des cellules pulmonaires, la fusion des MVB avec la membrane plasmique entraîne la libération de surfactant.
Récemment, Yefimova et al. ont mis en évidence la sécrétion de myélinosomes par des cellules de Sertoli en culture (la lignée murine de cellules TM4) [13, 14], des cellules nourricières qui interagissent de façon très étroite avec le gamète mâle avant sa libération dans le tube séminifère [15]. À ce niveau, les myélinosomes matures sont libérés en intégralité dans les espaces extracellulaires. Ce processus ressemble à l’exocytose impliquant les MVB qui conduit à la libération d’exosomes. D’ailleurs, myélinosomes et exosomes sont co-transportés au sein des MVB [13, 14] (Figure 2).
Les myélinosomes, un nouveau type de vésicules extracellulaires
Les vésicules extracellulaires (VE) sont des organites constituées d’une bicouche lipidique délimitant un contenu varié de protéines solubles ou membranaires, de lipides, de métabolites et d’acides nucléiques. Elles dérivent de membranes cellulaires libérées dans le milieu extracellulaire et sont présentes dans les fluides de l’organisme (sang, urine, lymphe, liquide céphalo-rachidien, plasma séminal, etc.). Hétérogènes en taille et en contenu, on en distingue deux sous-types : les microvésicules (MV) et les exosomes. Les myélinosomes font partie de ces VE [16]. Il est en effet possible de les isoler par centrifugation différentielle et de les concentrer avec les MV à 20 000 × g, les exosomes étant obtenus à 100 000 × g. La microscopie électronique reste la technique de référence pour déterminer la morphologie des VE [17]. Les exosomes et les MV peuvent être distingués par leur signature protéique spécifique qui dépend de leur origine subcellulaire différente [18]. De par leur biogenèse, les exosomes sont ainsi enrichis en protéines impliquées dans la sortie endosomale, comme les tétraspanines (Tsg101 [tumor susceptibility gene 101], CD9, CD63) ou Alix (ALG-2 interacting protein X). Certaines familles de protéines sont néanmoins présentes dans les deux sous-types de vésicules, comme des protéines du cytosquelette ou de la matrice extracellulaire, du complexe majeur d’histocompatibilité, des chaperonnes, des protéines liant les acides nucléiques et des protéines impliquées dans le trafic membranaire [19] (→).
(→) Voir la Nouvelle de J. Puyal et al., m/s n° 1, janvier 2008, page 19
(→) Voir la Synthèse de A. Buschiazzo et al., m/s n° 11, novembre 2019, page 852
(→) Voir la Synthèse de S. Le Lay et al., m/s n° 11, novembre 2018, page 936
Les VE ont la capacité de transmettre des informations aux cellules qui les reçoivent, en particulier celles qui sont associées à la biologie de la reproduction, par le biais du transfert de protéines fonctionnelles ou d’informations génétiques. Les VE du plasma séminal participent ainsi à divers aspects de la fertilité masculine, en améliorant la capacitation des spermatozoïdes, en induisant la réaction acrosomique, en stimulant la motilité des spermatozoïdes. Les myélinosomes diffèrent des exosomes et des microvésicules de par leur morphologie, leur origine et leur composition. La protéine CD63, caractéristique des exosomes, en est par exemple absente (Figure 3). Ils constituent donc un troisième sous-type de VE dont nous avons montré la présence dans le plasma séminal humain [16]. Cependant, le rôle physiologique des myélinosomes dans le liquide séminal humain reste à établir.
 |
Figure 3. Production des différents types de vésicules extracellulaires par la cellule. Les myélinosomes sont produits dans des situations pathologiques ou de stress en lien avec des facteurs génétiques ou environnementaux. |
Implication des myélinosomes dans les maladies d’agrégation et lysosomales
L’oligomérisation, puis la fibrillation de protéines mal repliées, caractérise un groupe de maladies appelées maladies d’agrégation des protéines. Certaines de ces altérations s’accompagnent d’une atteinte neurodégénérative et provoquent des lésions irréversibles du système nerveux central. Parmi les maladies neurodégénératives héréditaires, la maladie de Huntington (MH) est bien caractérisée. Elle est causée par une expansion de triplets CAG dans le gène codant la protéine Huntingtine (HTT) qui génère une longue chaîne de polyglutamines dans la séquence de la HTT, entraînant la formation d’agrégats insolubles. La présence de myélinosomes intracellulaires a été suggérée dans des neurones striataux de souris-modèles de maladie de Huntington [20].
La mucoviscidose (MCV) est une autre maladie d’agrégation. La forme la plus fréquente de la maladie est liée à la délétion de trois nucléotides du gène codant la protéine CFTR (cystic fibrosis transmembrane conductance regulator), un canal ionique régulant le transport des ions chlorures à travers la membrane plasmique de la cellule. Cette délétion est à l’origine de la disparition de la phénylalanine en position 508 de la protéine [21, 22] qui prédispose la protéine F508delCFTR mutée à l’agrégation [23]. Des myélinosomes intracellulaires ont été détectés dans les cellules alvéolaires pulmonaires, le poumon étant un organe très affecté dans cette maladie [24]. Là encore, leur signification fonctionnelle reste encore à établir.
Une complication majeure de la maladie de Fabry, une anomalie de stockage lysosomal liée à l’X résultant d’un défaut d’activité de l’α-galactosidase, est une néphropathie se traduisant par une perte progressive de la fonction rénale. Dans cette maladie également, des myélinosomes ont été observés dans le parenchyme rénal de patients, sans que leur fonction soit élucidée [25, 26].
Malgré le caractère ubiquitaire de l’agrégation de la HTT mutée dans tous les organes des patients et chez les animaux-modèles atteints de maladie de Huntington, certaines cellules « privilégiées » échappent naturellement à l’agrégation. Parmi ces cellules « privilégiées », on trouve les kératinocytes et les cellules testiculaires germinales et somatiques [27]. Dans le cas de la mucoviscidose, les cellules « privilégiées » sont, dans l’épithélium broncho-alvéolaire, des cellules sécrétoires responsables de la production de surfactant, contrairement aux cellules ciliées qui sont altérées [24]. Ainsi, que ce soit dans la maladie de Huntington ou dans la mucoviscidose, les cellules « privilégiées » présentent une activité sécrétoire à l’origine d’un micro-environnement spécifique des différents tissus (cellules de Sertoli à partir de tubes séminifères, cellules pulmonaires alvéolaires de type II), ou subissent un important remaniement de structure, avec la perte de cytoplasme au cours du processus de différenciation (les cellules germinales se différenciant d’une spermatide ronde à un spermatozoïde mature). Les cellules de Sertoli, comme probablement d’autres cellules « privilégiées », utilisent la voie des myélinosomes pour maintenir leur protéostase et leur survie, évitant ainsi l’accumulation de protéines potentiellement toxiques et de métabolites non dégradés qui auraient tendance à s’agréger. Nous avons en effet détecté des myélinosomes dans différentes lignées cellulaires, telles que les cellules gliales rétiniennes, les cellules broncho-épithéliales modèle de mucoviscidose ou les cellules de neuroblastome humain (Yefimova et al., manuscrit en préparation).
Gestion non catabolique des protéines mal repliées : un nouvel aspect du contrôle de qualité des protéines
La sécrétion de myélinosomes, ou voie MDS pour myelinosome-driven secretion, est une nouvelle stratégie non catabolique de contrôle de qualité des protéines qui coexiste avec les mécanismes de dégradation classiques, tels que la dégradation par le protéasome ou par l’autophagie (Figure 1). D’autres mécanismes de sécrétion non conventionnelle ont également été caractérisés, permettant à des protéines eucaryotes dépourvues de séquence signal de sortir de la cellule [6, 28]. Ainsi, la sécrétion de protéines associée à un repliement anormal (misfolding-associated protein secretion, MAPS) est un mécanisme qui a récemment été décrit : il cible les protéines cytosoliques mal repliées vers une voie de sécrétion non conventionnelle [6]. Dans ce processus, les protéines ubiquitinylées, normalement destinées au protéasome, subissent une dé-ubiquitination par la USP19 (ubiquitin-specific peptidase 19) qui les associe ensuite au réticulum endoplasmique. Ces protéines sont insérées dans des endosomes et sont sécrétées sous forme libre dans l’espace extracellulaire lorsque ceux-ci fusionnent avec la membrane plasmique [6, 28]. Dans la voie MDS, les proteines mal repliées ne sont pas libérées mais orientées vers des myélinosomes.
Les myélinosomes sont produits par la cellule dans des situations pathologiques ou de stress en lien avec des facteurs génétiques ou environnementaux. Dans les maladies d’agrégation telles que la maladie de Huntington ou la mucoviscidose, les myélinosomes semblent être des organites protecteurs préservant les cellules des protéines potentiellement toxiques par agrégation tout en maintenant la protéostase. La présence dans l’espace extracellulaire de myélinosomes comportant des protéines mal repliées pose la question de la possible transmission de cellule à cellule de ces protéines anormales. Des myélinosomes ont été observés dans la maladie de Huntington, mais également dans la maladie de Parkinson ou la sclérose latérale amyotrophique (SLA). Ils pourraient jouer un rôle délétère dans l’évolution de ces maladies neuro-dégénératives. Les myélinosomes pourraient par ailleurs jouer un rôle dans le transfert d’informations au niveau du liquide séminal. Cette activité pourrait représenter une nouvelle approche et un défi pour l’amélioration de la qualité des spermes humains.
Nouveaux types de vésicules extracellulaires, les myélinosomes présentent également un potentiel non encore exploré de biomarqueurs prédictifs de pathologies. Leur présence dans les milieux extracellulaires in vivo est désormais avérée, et on les retrouve dans différents liquides biologiques, comme le liquide séminal [16]. La recherche de myélinosomes (et la caractérisation de leur contenu) au sein de différents fluides biologiques comme le sérum, le liquide céphalo-rachidien, les lavages broncho-alvéolaires ou le fluide folliculaire, pourraient constituer dans le futur un marqueur de l’évolution de certaines maladies génétiques liées à l’accumulation de protéines mal repliées.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Matériel supplémentaire
Figure (supplémentaire). Accéder au matériel supplémentaire
Stress protéotoxique : stress lié à la production trop importante de protéines mal repliées susceptibles de former des agrégats toxiques pour la cellule.
Osmiophile : caractéristique en microscopie électronique des membranes à double couche phospholipidique qui lient l’osmium utilisé comme produit de contraste.
Références
- Arrigo AP. Chaperons moléculaires et repliement des protéines : l’exemple de certaines protéines de choc thermique. Med Sci (Paris) 2005 ; 21 : 619–625. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Le Benaroudj N.. protéasome, une machinerie cellulaire qui dégrade les protéines. Med Sci (Paris) 2005 ; 21 : 115–116. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Puyal J, Ginet V, Vaslin A, Clarke P. L’autophagie remplaçant de luxe du protéasome. Med Sci (Paris) 2008 ; 24 : 19–21. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Buschiazzo A, Yefimova M, Bourmeyster N, et al. Autophagie et spermatozoïde. Med Sci (Paris) 2019 ; 35 : 852–858. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Wosen JE, Mukhopadhyay D, Macaubas C, Mellins ED. Epithelial MHC class II expression and its role in antigen presentation in the gastrointestinal and respiratory tracts. Front Immunol 2018 ; 9 : 2144. [CrossRef] [PubMed] [Google Scholar]
- Lee JG, Takahama S, Zhang G, et al. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat Cell Biol 2016 ; 18 : 765–776. [CrossRef] [PubMed] [Google Scholar]
- Rabouille C.. Pathways of unconventional protein secretion. Trends Cell Biol 2017 ; 27 : 230–240. [Google Scholar]
- Ghadially FN. UItrastructural pathology of the cell and matrix. 3rd ed. Londres : Butterworths, 1989. [Google Scholar]
- Prince JS, Kohen C, Kohen E, et al. Direct connection between myelinosomes, endoplasmic reticulum and nuclear envelope in mouse hepatocytes grown with the amphiphilic drug, quinacrine. Tissue Cell 1993 ; 25 : 103–110. [CrossRef] [PubMed] [Google Scholar]
- Platt FM, Boland B, van der Spoel AC. The cell biology of disease lysosomal storage disorders the cellular impact of lysosomal dysfunction. J Cell Biol 2012 ; 199 : 723–734. [CrossRef] [PubMed] [Google Scholar]
- Marks MS, Heijnen HF, Raposo G. Lysosome-related organelles: unusual compartments become mainstream. Curr Opin Cell Biol 2013 ; 25 : 495–505. [CrossRef] [PubMed] [Google Scholar]
- Whitsett JA, Weaver TE. Alveolar development and disease. Am J Respir Cell Mol Biol 2015 ; 53 : 1–7. [CrossRef] [PubMed] [Google Scholar]
- Yefimova MG, Béré E, Cantereau-Becq A, et al. Myelinosomes act as natural secretory organelles in Sertoli cells to prevent accumulation of aggregate-prone mutant Huntingtin and CFTR. Hum Mol Genet 2016 ; 25 : 4170–4185. [CrossRef] [PubMed] [Google Scholar]
- Yefimova M, Bourmeyster N. Myelinosome-driven secretion: non catabolic management of misfolded proteins-Lessons from the Sertoli cells. J Rare Dis Res Treat 2017 ; 2 : 24–27. [Google Scholar]
- Ravel C, Jaillard S. The Sertoli cell. Morphologie 2011 ; 95 : 151–158. [CrossRef] [PubMed] [Google Scholar]
- Yefimova M, Bere E, Neyroud AS, et al. Myelinosome-like vesicles in human seminal plasma: a cryo-electron microscopy study. Cryobiology 2019 ; 2240 : 30168–30163. [Google Scholar]
- Coumans FAW, Brisson AR, Buzas EI, et al. Methodological guidelines to study extracellular vesicles. Circ Res 2017 ; 120 : 1632–1648. [PubMed] [Google Scholar]
- Kowal J, Arras G, Colombo M, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA 2016 ; 113 : E968–E977. [CrossRef] [PubMed] [Google Scholar]
- Le Lay S, Martinez MC, Andriantsitohaina R. Vésicules extracellulaires, biomarqueurs et bioeffecteurs du syndrome métabolique. Med Sci (Paris) 2018 ; 34 : 936–943. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Chen J, Marks E, Lai B, et al. Iron accumulates in Huntington’s disease neurons: protection by deferoxamine. PLoS One 2013 ; 8 : e7702. [Google Scholar]
- Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989 ; 245 : 1066–1073. [Google Scholar]
- Yefimova M, Bourmeyster N, Becq F, et al. Update on the cellular and molecular aspects of cystic fibrosis transmembrane conductance regulator (CFTR) and male fertility. Morphologie 2019 ; 103 : 4–10. [CrossRef] [PubMed] [Google Scholar]
- Kopito R.. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 2000 ; 10 : 524–530. [Google Scholar]
- Du K, Karp PH, Ackerley C, et al. Aggregates of mutant CFTR fragments in airway epithelial cells of CF lungs: new pathologic observations. J Cyst Fibros 2015 ; 14 : 182–193. [PubMed] [Google Scholar]
- Torra R, Algaba F, Ars E, et al. Preservation of renal function in a patient with Fabry nephropathy on enzyme replacement therapy. Clin Nephrol 2008 ; 69 : 445–449. [CrossRef] [PubMed] [Google Scholar]
- Fervenza FC, Torra R, Warnock DG. Safety and efficacy of enzyme replacement therapy in the nephropathy of Fabry disease. Biologics 2008 ; 2 : 823–843. [PubMed] [Google Scholar]
- Sathasivam K, Hobbs C, Turmaine M, et al. Formation of polyglutamine inclusions in non-CNS tissue. Hum Mol Genet 1999 ; 8 : 813–822. [CrossRef] [PubMed] [Google Scholar]
- Malhotra V.. Unconventional protein secretion: an evolving mechanism. EMBO J 2013 ; 32 : 1660–1664. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Différents mécanismes de contrôle de la qualité des protéines. Les protéines mal repliées sont repérées par différents mécanismes de contrôle comme le système ubiquitine/protéasome, l’autophagie (macroautophagie ou autophagie liée aux chaperonnes) ou le mécanisme MAPS (misfolded-associated protein secretion). Un nouveau mécanisme consiste en l’incorporation des protéines mal repliées au sein de myélinosomes qui sont ensuite éliminés par sécrétion extracellulaire (MDS). En cas d’échec ou de débordement de ces mécanismes de contrôle, les protéines anormales s’accumulent et forment des agrégats toxiques. |
| Dans le texte | |
|
Figure 2. Ultrastructure des myélinosomes. Photographie en microscopie électronique de milieu de culture de cellules de Sertoli. On constate au-delà des myélinosomes (voir insert) la présence de nombreuses autres vésicules extracellulaires (microvésicules et exosomes). Insert : à fort grossissement, on constate la présence de nombreuses membranes superposées entourant une matrice contenant une structure lucide aux électrons ainsi qu’une forme de cloisonnement. |
| Dans le texte | |
|
Figure 3. Production des différents types de vésicules extracellulaires par la cellule. Les myélinosomes sont produits dans des situations pathologiques ou de stress en lien avec des facteurs génétiques ou environnementaux. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.