")
")
| Issue |
Med Sci (Paris)
Volume 35, Number 8-9, Août–Septembre 2019
|
|
|---|---|---|
| Page(s) | 659 - 666 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2019128 | |
| Published online | 18 September 2019 | |
Syndrome néphrotique idiopathique et facteurs circulants
Une Arlésienne ?
Idiopathic nephrotic syndrome: une Arlésienne?
1
Inserm U1197, Interactions cellules souches-niches-physiologie, tumeurs et réparations tissulaires, Hôpital Paul Brousse, Bâtiment Lavoisier, 14, avenue Paul-Vaillant Couturier, 94800 Villejuif, France.
2
Université Paris-Saclay, Campus universitaire d’Orsay, 91 405 Orsay, France.
3
Service de néphrologie, CHU Bicêtre, 94270 Le Kremlin Bicêtre, France.
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
La fonction d’excrétion du rein fait intervenir des glomérules chargés de filtrer sélectivement le sang. L’acteur principal du filtre glomérulaire est le podocyte dont les pédicelles entrelacés portent des complexes moléculaires (néphrine, podocine, etc.) qui sont responsables du fonctionnement de la barrière de filtration (diaphragme de fente). Des altérations de ces podocytes entraînent une protéinurie massive qui caractérise le syndrome néphrotique. Parmi les formes les plus malignes de cette pathologie, se trouve le syndrome néphrotique idiopathique dont la physiopathologie reste inconnue. Ce syndrome regroupe essentiellement deux entités : les lésions glomérulaires minimes et la hyalinose segmentaire et focale. Ces pathologies impliqueraient les cellules du système immunitaire et plusieurs facteurs de perméabilité circulants qui agiraient sur la morphologie et le fonctionnement des podocytes.
Abstract
The renal filtration is ensured by the kidney glomeruli selective for filtering the blood. The main actor of the glomerular filter is the podocyte whose interlaced pedicels bear protein complexes (nephrin, podocin, etc.) creating a molecular sieve (slit diaphragm) to achieve the filtration. Alterations of these podocytes lead to massive proteinuria, which characterizes the nephrotic syndrome. The idiopathic form is one of the most malignant and essentially comprises two entities: minimal change disease and focal segmental glomerulosclerosis. Many observations indicated that (1) immune cells are involved and that (2) there are several permeability factors in the blood that affect the morphology and function of podocytes (slit diaphragm with fractional foot processes fusion/effacement). Evidence for a permeability factor was chiefly derived from remission of proteinuria observed after implantation of a kidney with FSGS in healthy recipients or with other kidney diseases. Today, we are moving towards a multifactorial conception of the nephrotic syndrome where all these barely known factors could be associated according to a sequential kinetic mechanism that needs to be determined.
© 2019 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (http://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (http://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Les reins représentent 1 % du poids du corps mais 10 % de la consommation totale d’oxygène. C’est un organe complexe, tant des points de vue de son développement, de sa structure, de sa physiologie que de sa pathologie. L’une des pathologies qui le touche, le syndrome néphrotique, a été décrite pour la première fois par Hippocrate comme un œdème généralisé. Il faudra attendre 1722 et Théodore Zwinger pour que le rein fasse son entrée dans cette pathologie. On s’aperçut ensuite que les urines des patients atteints de cette maladie pouvaient coaguler, et c’est Richard Bright, en 1827, qui, pour la première fois, a pu rassembler la triade : œdème généralisé, protéinurie et maladie rénale.
Le développement rénal
C’est chez les mammifères que le développement rénal prend toute son ampleur (Figure 1). On observe initialement, selon une organisation spatio-temporelle précise, un rein céphalique, nommé pronéphros. Ce rein est à l’origine de la formation du canal de Wolff qui induira, le long de son parcours dans le mésoderme intermédiaire, le développement du rein thoracique encore nommé mésonéphros. Le canal de Wolff continue sa progression vers la partie postérieure du fœtus humain. Il émettra au cours de la cinquième semaine de gestation, un bourgeon inducteur (bourgeon urétéral) qui induira dans le blastème métanéphrogène la formation du rein définitif des mammifères : le métanéphros. C’est dans ce rein que se différencient les unités filtrantes que l’on appelle néphrons. Il y a environ 900 000 néphrons par rein, mais ce nombre varie en fonction du développement intra-utérin, de l’environnement du fœtus et de son poids à la naissance. Un faible poids de naissance est en effet associé à une réduction du nombre de néphrons par rein, qui devient alors plus sensible aux maladies rénales aux cours de sa vie [1].
 |
Figure 1. Cinétique du développement rénal chez l’homme. |
Des mutations, impliquant actuellement plus de 50 gènes, peuvent perturber le développement et la différenciation du rein, en particulier au niveau de la barrière de filtration et des podocytes. Certains des allèles mutés sont à l’origine d’une protéinurie in utero ou/et de la formation d’un rein plus petit que la normale [2].
Avec l’âge, un déclin de la taille totale des néphrons et de leur nombre apparaît. Des changements tubulo-interstitiels, un épaississement de la membrane basale glomérulaire et une augmentation de la glomérulosclérose sont aussi observés [3]. On note ainsi une « certaine ressemblance » entre population âgée en bonne santé et population qui présente une maladie rénale chronique.
Structure du rein
Chaque néphron reçoit le sang par une artériole afférente (subdivision terminale de l’artère rénale), qui forme un peloton vasculaire appelé glomérule. Le sang repart ensuite par une artériole efférente (Figure 2A). Les cellules endothéliales de ce glomérule présentent un cytoplasme fenestré et l’endothélium est recouvert d’un glycocalyx dense. Il filtre sélectivement le sang. Le glomérule est enveloppé par la capsule de Bowman1, l’ensemble constituant le corpuscule rénal. Le versant externe de cette capsule est constitué d’un épithélium pariétal qui fait face à un épithélium viscéral, composé de cellules originales - les podocytes - qui déterminent en grande partie la capacité de filtration de cet organe. Chez l’adulte, on compte entre 500 et 600 podocytes par corpuscule. Ils possèdent des pédicelles enchevêtrés les uns dans les autres, ne laissant qu’un espace de 40 nm entre eux, et sont recouverts de molécules spécialisées dans l’adhérence cellulaire (podocine, néphrine, etc.). Cet espace constitue ce que l’on nomme le diaphragme de fente (Figures 2B, C et 3). Ces podocytes sont doués de très nombreuses propriétés. Ils ont en effet des propriétés de cellules présentatrices d’antigènes, d’endocytose, de contraction (via l’actine du cytosquelette), d’adhérence à la membrane basale glomérulaire (en particulier par les intégrines a3b1 et avb3), de filtration sélective des molécules au niveau du diaphragme de fente, selon leur charge et leur taille (les protéines de masses moléculaires supérieures à 65-68 000 daltons - comme l’albumine – sont en effet arrêtées).
 |
Figure 2. A. Histologie d’un glomérule (Gl) sain chez l’homme (coloration hémalun/éosine x630). Le sang arrive au Gl par l’artériole afférente, il est distribué aux capillaires glomérulaires soutenus par les cellules mésangiales et la matrice qu’elles produisent. Le cytoplasme fenestré des cellules endothéliales des capillaires est orienté vers la membrane basale glomérulaire (MBG) localisée directement à leur contact. Les podocytes reposent sur cette MBG. L’ultrafiltration du sang à travers ces différentes barrières donne naissance à l’urine primitive déversée dans l’espace urinaire et recueillie au pôle tubulaire à l’opposé du pôle vasculaire. L’ultrafiltat sera évacué par le tube contourné proximal. Le sang quitte le Gl par l’artériole efférente. B. Les pédicelles enchevêtrés des podocytes de l’épithélium viscéral de la capsule de Bowman sont en contact avec la MBG et délimitent entre eux l’espace de filtration ou diaphragme de fente. L’autre versant de la MBG est tapissé par l’endothélium du Gl qui limite l’interface sanguine. C. Représentation de quelques molécules qui interviennent au niveau du diaphragme de fente et de la surface basale des podocytes. Le composant majeur du diaphragme est la néphrine. Ces molécules présentent des interactions entre elles et une molécule en forme de harpon: la podocine. Ce complexe moléculaire est lié au cytosquelette d’actine par un complexe intracytoplasmique (non détaillé) déterminé par la protéine associée à CD2 (CD2AP). Le réseau d’actine est régulé en particulier par l’a-actinine-4 mais aussi par la synaptopodine. Au niveau de la surface basale, les molécules d’adhérence telles que l’intégrine a3b1 et l’a-dystroglycane sont liées à la laminine tandis que l’intégrine avb3 est associée à la vitronectine dans la MBG. C’est la sous-unité b des intégrines qui intervient. Ces intégrines sont couplées au réseau d’actine par le complexe taline/vinculine/paxilline tandis que l’a/b-dystroglycane est liée à l’actine par la molécule d’urotrophine. Les podocytes expriment à leur surface des récepteurs d’interleukines et les molécules CD40 et CD80. Ils sont aussi capables d’endocytose. |
 |
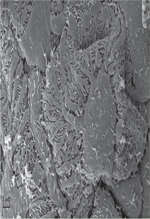
Figure 3. Microscopie électronique à balayage d’un rein normal montrant les podocytes avec leurs pédicelles enchevêtrés et les diaphragmes de fente. Comparer avec la Figure 2B. |
Les podocytes sont ancrés, via leurs pédicelles, dans la membrane basale des glomérules, synthétisée par les podocytes eux-mêmes (collagène IV sous-unités a3,4,5 disposé au centre de cette membrane) et par les cellules endothéliales (collagène IV sous-unités a1,2 disposé du côté endothélial de la basale). Cette structure est associée à d’autres molécules, comme les protéoglycanes, qui lui confèrent des propriétés anioniques responsables de la sélectivité de charge lors de la filtration rénale. Le filtrat glomérulaire passe ainsi, à partir du sang artériel, au travers de l’ensemble de ces tissus, et atteint la chambre urinaire, délimitée par la capsule de Bowman. On parle alors d’urine primitive (Figure 2A, B). Un tissu de soutien, appelé mésangium, constitué de cellules mésangiales, tapisse l’intérieur des glomérules. Ces cellules sont capables de se contracter. Elles produisent de la matrice extracellulaire et sont douées de phagocytose.
La physiologie rénale
Deux activités caractérisent la physiologie rénale. L’une contrôle, par les mécanismes de filtration glomérulaire et de réabsorption/sécrétion au niveau de différentes régions des néphrons, la concentration minérale du sang, l’équilibre acido/basique, l’excrétion des déchets solubles et le volume sanguin. L’autre repose sur la capacité du rein à fonctionner comme une glande hormonale. Le rein produit en effet l’érythropoïétine, qui est nécessaire à la synthèse des globules rouges, la 1-25 (OH)2 vitamine D, qui contrôle la concentration sanguine de calcium, et la rénine, impliquée dans la régulation de la pression artérielle. Ce petit organe de 12 cm joue donc de très nombreux rôles dans la physiologie générale et les 14 (au moins) types cellulaires différents constitutifs d’un néphron sont sollicités afin d’établir l’homéostasie nécessaire à la vie de l’organisme, quelles que soient les variations d’environnement auxquelles il est confronté.
Les pathologies rénales
Les pathologies touchant le rein sont classées selon les différentes structures histologiques qu’elles altèrent : glomérule/corpuscule, tubules, tissu interstitiel, vaisseaux. C’est environ 10 % de la population qui est affectée par une maladie rénale chronique et, pour les individus de 65 ans et plus, la prévalence est de 20 % [4]. Le risque d’évoluer vers la phase terminale reste inférieur à 1 % chez les patients de moins de 40 ans mais supérieur ou égal à 8 % chez les patients âgés de 65 ans et plus. Nous nous attacherons ici à développer les atteintes glomérulaires et plus particulièrement le syndrome néphrotique idiopathique (SNI) qui comporte essentiellement deux entités, les lésions glomérulaires minimes (LGM ou minimal change disease) et la hyalinose (ou glomérulosclérose) segmentaire et focale (HSF ou FSGS). En dépit de l’identification de nombreux facteurs (hypertension artérielle, hyperlipidémie, obésité, infections, médicaments, mutations géniques) qui peuvent conduire à la HSF, c’est approximativement 80 % des cas qui sont d’origine idiopathique [4]. Ces SNI représentent 15 à 30 % des glomérulopathies de l’adulte [5] et 85 à 90 % de celles de l’enfant [6] (→).
(→) Voir la Synthèse de V. Audard et al., m/s n° 10, octobre 2008, page 853, et la Nouvelle de S.Y. Zhang et al., m/s n° 6-7, juin-juillet 2010, page 592
Chez les enfants, l’incidence du syndrome néphrotique varie beaucoup selon le pays d’origine ou l’ethnie, de 1,15 à 16,9 pour 100 000 enfants [7].
Les lésions glomérulaires minimes
Les lésions glomérulaires minimes (LGM) représentent la cause la plus fréquente du syndrome néphrotique chez l’enfant (70 à 90 % des cas chez les moins de 10 ans et 50 % pour les plus âgés [8]), avec un pic de fréquence situé entre 2 et 6 ans. Mais les LGM sont aussi la cause de syndrome néphrotique chez les adultes pour 10 à 20 % des cas [9]. Elles sont caractérisées par une fusion et/ou l’effacement des pédicelles des podocytes, avec une hyperplasie des microvillosités, que l’on ne repère pas en microscopie optique, où les corpuscules rénaux paraissent normaux. Les anomalies ne sont observables qu’en microscopie électronique (Figure 4B). Ces lésions sont responsables principalement d’une protéinurie massive (plus de 3,5 g/jour chez l’adulte), en particulier d’une fuite de lipoprotéines, d’où l’autre appellation de cette affection, « néphrose lipoïdique », et d’un œdème interstitiel. La fonction rénale est préservée, on n’observe ni hypertension ni hématurie. Ces LGM répondent aux corticothérapies de façon spectaculaire (pour plus de 90 % des cas). Ainsi, en dépit des rechutes possibles, l’avenir de ces enfants est bon : le risque de progression vers une maladie rénale chronique, 10 ans après le diagnostic, est en effet estimé à moins de 5 % [10].
 |
Figure 4. A. Microscopie optique d’un rein présentant une HSF où l’on peut voir un glomérule abîmé par la fibrose (coloration au trichrome de Masson x10). B. Microscopie électronique d’une coupe de rein à LGM montrant l’aspect des pédicelles des podocytes. Dans différentes zones, ces pédicelles sont fusionnés. Cette photo montre aussi l’aspect de la MBG et de l’endothélium à cytoplasme fenestré des capillaires glomérulaires. GL : glomérulaire ; MBG : membrane basale glomérulaire. |
La hyalinose segmentaire et focale
Dans la hyalinose segmentaire et focale (HSF), la sclérose n’affecte qu’un secteur du glomérule, d’où le terme de « segmentaire », et seulement certains glomérules, ce qui explique le terme de « focal ». Ce syndrome néphrotique est directement observable en microscopie optique (Figure 4A). La classification de Columbia a établi 5 sous-types morphologiques de HSF selon la présentation histologique de l’anomalie glomérulaire [11]. Dans les glomérules atteints, les cellules mésangiales prolifèrent et le dépôt de matrice extracellulaire est plus ou moins important [12]. Les podocytes ont une structure altérée (leur réseau d’actine est modifié), leur nombre diminue - par mort cellulaire par apoptose/nécrose, ou migration par dissociation des complexes d’adhérence focaux et phosphorylation de VASP (vasodilator-stimulated-phosphoprotein). Les podocytes restants deviennent hypertrophiques, ce qui favorise le recouvrement de la surface des capillaires glomérulaires. Les corpuscules rénaux, moins nombreux, présentent, par adaptation, un plus grand diamètre associé à une adhérence plus ou moins importante du glomérule à la pariétale de la capsule (synéchie) aux niveaux laissés vacants après disparition des podocytes. Cette situation semble activer les cellules épithéliales pariétales de la capsule, qui peuvent se substituer aux podocytes perdus (rôle bénéfique), ou envahir le glomérule et sécréter de la matrice extracellulaire (rôle délétère) [13]. Un phénomène de fusion et/ou d’effacement des pédicelles des podocytes est aussi observé. Du point de vue clinique, ce syndrome néphrotique est caractérisé par une protéinurie abondante (supérieure à 3,5 g/24 h), un syndrome œdémateux et peut être associé à une hypoalbuminémie, une hyperlipidémie et une lipidurie. Le syndrome œdémateux correspond à un œdème interstitiel ; il est présent chez tous les individus atteints d’un syndrome néphrotique. Parmi les nombreuses protéines retrouvées dans les urines des patients, on observe des protéases dont certaines peuvent modifier les canaux sodium des épithéliums des tubes distaux et collecteurs. Ces canaux deviennent actifs, ce qui déclenche une forte réabsorption de sodium à ces niveaux. Chez le patient néphrotique, ce processus est l’une des explications à l’origine de la rétention importante de sodium responsable d’une augmentation du volume des fluides et d’une fuite du liquide en excès vers les tissus interstitiels (œdème). L’incidence de ce syndrome néphrotique, chez l’adulte, semble augmenter depuis les dernières décennies : elle oscillerait aujourd’hui entre 15 et 25 % [14]. Les HSF sont résistantes aux corticoïdes pour environ 70 % d’entre elles [15]. De manière générale, les syndromes néphrotiques idiopathiques résistants aux stéroïdes sont plus susceptibles de progresser vers une HSF et une insuffisance rénale terminale que les syndromes qui y sont sensibles [15]. C’est en particulier le cas chez les enfants, dont environ 50 % d’entre eux ont un risque de progresser vers une insuffisance rénale terminale [16].
Lésions glomérulaires minimes versus hyalinose segmentaire et focale
La perméabilité glomérulaire chez les patients atteints de hyalinose segmentaire et focale (HSF) est supérieure à celle des patients atteints de lésions glomérulaires minimes (LGM) et augmente avec la fréquence des rechutes [17]. Avec l’avènement de la biopsie, au milieu du XXe siècle, le développement de l’immunofluorescence et de la microscopie électronique, le diagnostic du syndrome néphrotique a été révolutionné. Des biopsies réalisées de façon répétée au cours de la maladie révèlent ainsi que les altérations des podocytes peuvent progresser des LGM jusqu’aux HSF récurrentes [18]. Tejani et al. [19] ont suivi des LGM avec de fréquentes rechutes en utilisant des biopsies réalisées chez des enfants, 1,5 an après le début de la maladie, puis 4,5 ans plus tard chez ceux qui souffrent de récidives. Les analyses histologiques de ces biopsies montrent que 55 % d’entre elles correspondent alors à des HSF. Un tel chevauchement entre LGM et HSF est également observé dans les modèles animaux. La sévérité des lésions est liée au degré de perte des podocytes : quand il est supérieur à 20 %, une protéinurie soutenue se développe, tandis qu’elle est transitoire si ce pourcentage est inférieur [12]. Les HSF seraient ainsi une conséquence de la diminution du nombre des podocytes, quand celle-ci dépasse un seuil critique. La cinétique d’évolution est donc la suivante : protéinurie, effacement des pédicelles, diminution du nombre de podocytes, adhérence du glomérule à la capsule de Bowman et, finalement, sclérose. Comme RJ Maas, on peut ainsi admettre que « minimal change disease and idiopathic HSF are manifestations of the same disease » [20]. Les deux syndromes avaient été distingués dans les années 1970, mais les travaux que nous venons de présenter suggèrent que LGM et HSF idiopathiques sont en fait des phénotypes différents d’une même maladie. Dans la suite de cet article, nous considérerons donc ces deux syndromes comme une progression dans le temps de la pathologie glomérulaire avec des lésions extensives des podocytes.
L’hypothèse des facteurs circulants
Historique
La responsabilité des facteurs circulants extrarénaux dans l’apparition des syndromes néphrotiques idiopathiques a été proposée pour la première fois par Gentili et al. en 1954. Une expérience particulièrement audacieuse, mais éthiquement discutable, a été réalisée par ces auteurs. En administrant du plasma d’enfants présentant une LGM à des enfants exempts de syndrome néphrotique, ils ont pu observer l’induction d’une légère protéinurie chez ces derniers ! [21]. Dans les années 1970, de nombreuses études cliniques font état de HSF récurrentes après des transplantations rénales. Hoyer et al., en 1972 [22], décrivent deux enfants et une femme présentant un syndrome néphrotique idiopathique résistant aux stéroïdes, qui, après deux à six ans, sont en insuffisance rénale terminale. Après transplantations, ces patients ont récidivé. Les auteurs remarquèrent que les altérations glomérulaires présentées par ces patients correspondaient à des HSF et commençaient à apparaître dans la zone de contact entre le cortex rénal et la médullaire, mais sans évidence, sur les biopsies, de signe de rejet. Cette cinétique médullo-corticale avait déjà été remarquée par Rich et al. en 1957 [23]. Ces résultats ont été le point de départ d’une réflexion sur l’implication de facteurs circulants à l’origine du syndrome néphrotique idiopathique [24]. Couser et Shankland font en effet remarquer que deux des trois patients suivis par Hoyer n’ont pas de sclérose initiale, et que les trois présentent une LGM sur les biopsies des transplants initiaux, au début de l’apparition du syndrome [24]. Il s’agit là d’un argument indiscutable indiquant que les facteurs circulants peuvent induire une LGM aussi bien qu’une HSF, et donc que ces deux lésions sont probablement liées pathogénétiquement. Cet argument a été renforcé par un cas de rémission de la protéinurie, après transplantation de reins d’un donneur présentant une LGM prouvée par biopsie, chez un patient exempt de LGM et HSF.
D’autres observations confirment la présence de facteurs circulants responsables des lésions glomérulaires et/ou podocytaires :
-
La rémission de la protéinurie par plasmaphérèse [25], ou immunoadsorption sur protéine A2 [26], ou sur anticorps anti-immunoglobulines humaines, ce qui suggère un lien entre facteurs circulants et cellules immunitaires [27].
-
Le sérum, le plasma ou des fractions de plasma de patients atteints de HSF récurrentes injectés dans l’aorte de rats induisent une protéinurie [28] et une augmentation de la perméabilité à l’albumine sur des glomérules isolés [29].
-
Les mères HSF peuvent transmettre la maladie à leurs enfants, et la protéinurie de ces enfants guérit spontanément en quelques semaines après l’accouchement [30].
• Chez un patient HSF qui a reçu un transplant rénal, une protéinurie massive s’est déclarée et n’a pas pu être soignée. Le greffon a alors été re-transplanté à un patient diabétique et cette nouvelle greffe a été un succès [31].
Le rôle des lymphocytes
Shalhoub et al. montrent en 1974 [32] pour la première fois que ces facteurs circulants peuvent être associés aux lymphocytes T (LT). Ces résultats découlent d’observations cliniques : (1) l’induction par le virus de la rougeole (à l’origine d’une déplétion des LT) d’une rémission spectaculaire du syndrome néphrotique - des patients qui présentaient des cas difficiles de syndrome ont ainsi été délibérément infectés par ce virus [33] ! ; (2) la thérapie utilisée contre certains lymphomes résout le syndrome néphrotique que présentent parfois ces patients ; (3) les rémissions du syndrome induites par les stéroïdes qui entraînent une déplétion préférentielle des LT.
En 1975, Lagrue et al. [34] montrèrent que les lymphocytes T produisent une cytokine qui agit comme un facteur de perméabilité des vaisseaux (VPF), et avec Shalhoub et al., furent les premiers à suggérer la présence de ces facteurs de perméabilité dans les LGM. Enfin, la cyclosporine A, qui est utilisée dans le traitement de la protéinurie, agit, pour partie, en induisant une immunosuppression via la calcineurine et son action sur les lymphocytes T.
Le rôle des lymphocytes B a été découvert fortuitement chez un patient traité par le rituximab (un anticorps anti-CD20) pour un purpura thrombocytopénique idiopathique, et chez qui une rémission de la protéinurie a été observée [35]. Le rituximab a également une action directe sur les podocytes : il réduit en effet la diminution de l’activité de la SMPDL3b (sphingomyelinase-like phosphodiesterase 3b) observée chez les patients présentant une hyalinose segmentaire et focale, ce qui stabilise le cytosquelette d’actine et la morphologie des podocytes, ceux-ci restant alors associés à la membrane basale glomérulaire et ne subissant plus l’apoptose [36].
Dans le cas du lymphome de Hodgkin, lorsqu’il est associé à un syndrome néphrotique idiopathique, une expression du gène c-mip (c-maf inducing protein) est observée dans les cellules tumorales mais aussi dans les podocytes. Cette double expression n’est pas observée en l’absence de syndrome néphrotique. Or, des souris transgéniques qui surexpriment le gène c-mip dans les podocytes développent une protéinurie massive sans lésions inflammatoire ni infiltration cellulaire. La protéine codée par c-mip interfère avec la signalisation du podocyte en inactivant la protéine tyrosine kinase Fyn qui ne phosphoryle plus la néphrine, ce qui conduit à une désorganisation du cytosquelette, un effacement des pédicelles et à une fuite importante de protéines [37].
Les récepteurs membranaires CD40 et CD80 (B7-1) présents sur les lymphocytes mais aussi sur les podocytes, ont été impliqués dans le syndrome néphrotique idiopathique. Les anticorps anti-CD40, présents dans le sérum des patients HSF et dans les biopsies glomérulaires, altèrent le cytosquelette des podocytes de ces patients et augmentent leur perméabilité glomérulaire [38]. L’expression de CD80 est plus élevée au cours des rechutes de LGM ; elle se normalise après rémission [39]. CD80, exprimé par les podocytes dans les biopsies de reins des patients LGM et HSF, favorise leur migration en inhibant leur adhérence résultant de la liaison de la taline à l’intégrine b1 (Figure 2C). L’abatacept, une protéine de fusion composée du domaine extracellulaire de CTLA-4 (cytotoxic T-lymphocyte antigen 4) fusionné à une région Fc d'IgG1 humaine, peut induire, en se liant à CD80, une rémission partielle ou totale de la HSF [40]. D’autres molécules ont été proposées pour être des facteurs circulants, comme l’interleukine 13 (IL-13) qui provoque chez le rat la fusion de 80 % des pédicelles et la diminution de l’expression par les podocytes des gènes codant la néphrine, la podocine, et le dystroglycane [41] ; la CLCF-1 (cardiotrophine-like cytokine-1), un membre de la famille des IL-6, qui diminue l’expression de la néphrine dans les glomérules [42] ; le TNF-a (tumor necrosis alpha) qui est en concentration élévée dans le sang des patients présentant un syndrome néphrotique et dont le taux se normalise après rémission [43] ; l’hémopexine (Hx), une protéine à activité sérine-protéase produite par le foie découverte dans les années 1980-1990 [44], qui modifie la composition de la membrane basale glomérulaire et l’organisation des filaments d’actine, dépendant de la néphrine, dans les podocytes. Jusqu’à présent, la présence d’hémopexine n’a été révélée que chez les patients LGM.
Les autres facteurs circulants
Nous avons découvert dans le sérum des patients HSF, la protéine CASK (ou kinase sérine/thréonine dépendante régulée par le complexe calcium/calmoduline) sécrétée par une catégorie de cellules sanguines (probablement les macrophages) et dont la présence, à partir d’une certaine concentration, modifie le réseau d’actine des podocytes, provoque une fusion de leurs pédicelles et une protéinurie [54]. Actuellement, la molécule réceptrice de l’urokinase (uPAR) suscite un grand intérêt. Il s’agit d’une glycoprotéine ancrée dans la membrane cellulaire des leucocytes (rarement les lymphocytes), des cellules endothéliales et des podocytes, par une structure glycosylphosphatidylinositol (GPI). Différentes enzymes peuvent cliver l’ancrage GPI de uPAR produisant la forme soluble de la protéine - suPAR- qui est alors libérée dans le sang [45]. suPAR induit un effacement des pédicelles, un détachement des podocytes de la membrane basale glomérulaire et une protéinurie via l’activation de l’intégrine avb3 associée à la vitronectine de la membrane basale glomérulaire [46] (Figure 2C). Un autre facteur a également été proposé : l’angiopoïétine-like 4. Elle est sécrétée par les podocytes sous une forme hyposialylée dont la surexpression a un effet protéinurique chez les LGM [47].
Les facteurs circulants : trésor ou méprise
Les différents facteurs proposés ne sont pas spécifiques des syndromes néphrotiques idiopathiques. suPAR peut être associée à de nombreuses maladies du foie, des infections, au lupus érythémateux disséminé, l’athérosclérose, etc., maladies qui ne comportent pas ou peu de protéinurie mais dans lesquelles sa concentration sérique est augmentée. Certains patients développent une HSF récurrente en l’absence de taux circulants de suPAR élévés. Il n’y a ainsi pas de corrélation directe entre le niveau circulant de ce facteur et l’intensité de la protéinurie [48]. On ne sait pas non plus si l’action de ces différents facteurs circulants dépend du type de HSF [49]. Autre difficulté, la plasmaphérèse réalisée chez les patients souffrant de HSF idiopathiques afin d’épurer leur sang, présente un taux de réussite variable [50]. Les facteurs circulants ne distinguent pas entre promoteurs et inhibiteurs de la perméabilité glomérulaire. Les facteurs inhibiteurs présents dans le sérum d’individus sains, peuvent être retrouvés dans l’urine de patients néphrotiques [51]. Ils pourraient se lier aux facteurs de perméabilité ou les dégrader par voie enzymatique. Par exemple, la fuite dans l’urine des inhibiteurs de l’activation de l’hémopexine, favoriserait l’apparition de sa forme active et le développement de la pathologie. Le rôle de ces facteurs circulants serait en partie secondaire à la protéinurie.
Les concentrations, en tenant compte de la valeur de la filtration glomérulaire, des facteurs circulants (comme suPAR ou l’hémopexine) ne sont pas significativement différentes chez les patients LGM et chez les patients HSF, voire même chez les individus sains [52]. Les rôles pathologiques de ces facteurs pourraient être associés à leur état de glycosylation à l’origine de caractéristiques fonctionnelles différentes (par exemple, la formation de dimères pour suPAR). Ces facteurs pourraient également favoriser l’influx de calcium au niveau des canaux TRPC-6 (transient receptor potential channel-6) des podocytes et ainsi provoquer l’activation de la calcineurine podocytaire responsable d’une déphosphorylation de la synaptopodine. Sous cette forme, la synaptopodine qui n’est plus protégée de l’action protéolytique de la cathepsine-L, sera dégradée. L’association entre synaptopodine et actine, nécessaire à la stabilité du réseau d’actine (Figure 2C), n’étant plus assurée, une protéinurie apparaît [53]. En conditions physiologiques, la protéinurie peut être ajustée afin, par exemple, d’augmenter la clairance d’antigènes délétères lors d’une infection ou d’épisodes allergiques. Cette réponse repose sur les lymphocytes T régulateurs, mais aussi sur les podocytes par l’intermédiaire des interleukines qu’ils produisent. L’ensemble de ces facteurs qui présentent chacun des caractéristiques originales, pourrait participer à un « ballet » moléculaire, dont chaque intervenant entrerait « dans la danse » avec une cinétique temporelle et spatiale particulières. Dans ce « ballet », se formeraient des complexes moléculaires transitoires, dont la localisation, la concentration, la durée de présence et le moment de leur sécrétion, conduiraient aux différents profils histo-pathologiques que l’on observe, amplifieraient leurs manifestations, voire rendraient les podocytes plus sensibles aux modifications de leur environnement (infections, médicaments, etc.).
Une équipe allemande [55] vient de publier un travail élégant fondé sur la construction d’une continuité de la circulation sanguine entre deux larves de poissons zebrafish (parabiose). Chez l’un des zebrafish, le gène de la néphronectine (un composant de la membrane basale glomérulaire) a été invalidé, ce qui induit une protéinurie. Chez l’autre poisson (normal) se développe une protéinurie par effacement des pédicelles des podocytes. La protéinurie induit donc la production de facteurs circulants qui sont passés dans le zebrafish non traité ou bien l’absence de facteurs indispensables perdus sous l’effet de cette protéinurie. Conclusion : « la protéinurie déclenche la protéinurie » par un phénomène de feedback positif et pourrait exacerber d’autres maladies rénales.
L’origine de ces pathologies rénales, suivant les formes différentes que prennent le syndrome néphrotique idiopathique et les multiples facteurs circulants responsables de son apparition, reste difficile à comprendre. Si les facteurs circulants peuvent modifier la structure des podocytes et ainsi conduire à des anomalies fonctionnelles responsables d’un syndrome néphrotique idiopathique, la cause initiale de ce syndrome pourrait être reliée à une réponse anormale des podocytes à des évènements déclencheurs communs, tels que des infections ou des inflammations. Il existe sans doute des caractéristiques intrinsèques aux podocytes qui favoriseraient leur vulnérabilité. Désormais, il semble que le mécanisme ait une origine multifactorielle et ne soit pas associé à une seule molécule circulant dans le plasma des patients. Ces facteurs circulants, dont on parle depuis plus de 40 ans, ressemblent de plus en plus à l’Arlésienne de Bizet, toujours attendue, jamais trouvée…
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Nous sommes redevables au Docteur Michaël Trichet (Institut de Biologie, Paris-Seine) pour son expertise et son amicale attention en microscopie électronique, à la Région Île-de-France, Sorbonne-Université et au CNRS, ainsi qu’au Professeur Djillali Sahali (Hôpital Henri Mondor, Créteil) pour sa relecture critique du manuscrit, et au Docteur Sophie Ferlicot (CHU Bicêtre, Anatomie-Pathologique) pour sa participation à l’iconographie de l’HSF. Nous remercions Madame Véronique Mamessier pour son aide en histologie.
Ce travail est supporté par la Fondation du rein sous égide de la Fondation pour la recherche médicale. Grant numéro R16099LL de H.K. Lorenzo.
Décrite par William Bowman (1816-1892).
La protéine A est une protéine bactérienne fixant les immunoglobulines d’isotype G (IgG). Elle a ici la même utilisation que les anticorps anti-immunoglobuline.
Références
- Puelles VG, Hoy WE, Hughson MD, et al. Glomerular number and size variability and risk for kidney disease. Curr Opin Nephrol Hypertens 2011 ; 20 : 7–15. [CrossRef] [PubMed] [Google Scholar]
- Cil O, Perwad F. Monogenic causes of proteinuria in children. Front Med 2018 ; 5 : 55. [Google Scholar]
- O’Sullivan ED, Hughes J, Ferenbach DA. Renal aging: causes and consequences. J Am Soc Nephrol 2017 ; 28 : 407–420. [Google Scholar]
- D’Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med 2011 ; 365 : 2398–2411. [Google Scholar]
- Audard V, Lang P, Sahali D. Pathogénie du syndrome néphrotique à lesions glomérulaires minimes. Med Sci (Paris) 2008 ; 24 : 853–858. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Zhang SY, Audard V, Lang P, Sahali D. Mécanismes moléculaires du syndrome néphrotique idiopathique à rechutes: rôle de c-mip dans les dysfonctions podocytaires. Med Sci (Paris) 2010 ; 26 : 592–596. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Banh TH, Hussain-Shamsy N, Patel V, et al. Ethnic differences in incidence and outcomes of childhood nephrotic syndrome. Clin J Am Soc Nephrol 2016 ; 11 : 1760–1768. [CrossRef] [PubMed] [Google Scholar]
- Meyrier A, Niaudet P. Acute kidney injury complicating nephrotic syndrome of minimal change disease. Kidney Int 2018 ; 94 : 861–869. [CrossRef] [PubMed] [Google Scholar]
- Uezono S, Hara S, Sato Y, et al. Renal biopsy in elderly patients: a clinicopathological analysis. Renal Failure 2006 ; 28 : 549–555. [CrossRef] [PubMed] [Google Scholar]
- Mendonca AC, Oliveira EA, Froes BP, et al. A predictive model of progressive chronic kidney disease in idiopathic nephrotic syndrome. Pediatr Nephrol 2015 ; 30 : 2011–2020. [CrossRef] [PubMed] [Google Scholar]
- Han MH, Kim YJ. Practical application of Columbia classification for focal segmental glomerulosclerosis. BioMed Res Int 2016 ; 2016 : 9375753. [Google Scholar]
- Wharram BL, Goyal M, Wiggins JE, et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 2005 ; 16 : 2941–2952. [Google Scholar]
- Smeets B, Kuppe C, Sicking EM, et al. Parietal epithelial cells participate in the formation of sclerotic lesions in focal segmental glomerulosclerosis. J Am Soc Nephrol 2011 ; 22 : 1262–1274. [Google Scholar]
- O’Shaughnessy MM, Hogan SL, Poulton CJ, et al. Temporal and demographic trends in glomerular disease epidemiology in the southeastern United States, 1986–2015. Clin J Am Soc Nephrol 2017 ; 12 : 614–623. [CrossRef] [PubMed] [Google Scholar]
- Savin VJ, Sharma M, Zhou J, et al. Multiple targets for novel therapy of FSGS associated with circulating permeability factor. BioMed Res Int 2017 ; 2017 : 6232616. [Google Scholar]
- Ehrich JH, Geerlings C, Zivicnjak M, et al. Steroid-resistant idiopathic childhood nephrosis: overdiagnosed and undertreated. Nephrol Dial Transplant 2007 ; 22 : 2183–2193. [CrossRef] [PubMed] [Google Scholar]
- Sharma M, Zhou J, Gauchat JF, et al. Janus kinase 2/signal transducer and activator of transcription 3 inhibitors attenuate the effect of cardiotrophin-like cytokine factor 1 and human focal segmental glomerulosclerosis serum on glomerular filtration barrier. Transl Res 2015 ; 166 : 384–398. [Google Scholar]
- Fogo AB. Causes and pathogenesis of focal segmental glomerulosclerosis. Nat Rev Nephrol 2015 ; 11 : 76–87. [CrossRef] [PubMed] [Google Scholar]
- Tejani A, Phadke K, Nicastri A, et al. Efficacy of cyclophosphamide in steroid-sensitive childhood nephrotic syndrome with different morphological lesions. Nephron 1985 ; 41 : 170–173. [CrossRef] [PubMed] [Google Scholar]
- Maas RJ, Deegens JK, Smeets B, et al. Minimal change disease and idiopathic FSGS: manifestations of the same disease. Nat Rev Nephrol 2016 ; 12 : 768–776. [CrossRef] [PubMed] [Google Scholar]
- Gentili A, Tangheroni W, Gelli G. Proteinuria caused by transfusion of blood from nephrotic to non-nephrotic individuals. Minerva Medica 1954 ; 45 : 603–608. [PubMed] [Google Scholar]
- Hoyer JR, Vernier RL, Najarian JS, et al. Recurrence of idiopathic nephrotic syndrome after renal transplantation. Lancet 1972 ; 2 : 343–348. [CrossRef] [PubMed] [Google Scholar]
- Rich AR. A hitherto undescribed vulnerability of the juxtamedullary glomeruli in lipoid nephrosis. Bull Johns Hopkins Hosp 1957 ; 100 : 173–186. [PubMed] [Google Scholar]
- Hoyer JR, Vernier RL, Najarian JS, et al. Recurrence of idiopathic nephrotic syndrome after renal transplantation. 1972. J Am Soc Nephrol 2001 ; 12 : 1994–2002. [Google Scholar]
- Dantal J, Testa A, Bigot E, Soulillou JP. Effects of plasma-protein A immunoadsorption on idiopathic nephrotic syndrome recurring after renal transplantation. Ann Med Interne (Paris) 1992 ; 143 : suppl 1 48–51. [PubMed] [Google Scholar]
- Artero ML, Sharma R, Savin VJ, Vincenti F. Plasmapheresis reduces proteinuria and serum capacity to injure glomeruli in patients with recurrent focal glomerulosclerosis. Am J Kidney Dis 1994 ; 23 : 574–581. [CrossRef] [PubMed] [Google Scholar]
- Dantal J, Godfrin Y, Koll R, et al. Antihuman immunoglobulin affinity immunoadsorption strongly decreases proteinuria in patients with relapsing nephrotic syndrome. J Am Soc Nephrol 1998 ; 9 : 1709–1715. [Google Scholar]
- Zimmerman SW. Increased urinary protein excretion in the rat produced by serum from a patient with recurrent focal glomerular sclerosis after renal transplantation. Clin Nephrol 1984 ; 22 : 32–38. [PubMed] [Google Scholar]
- Savin VJ, Sharma R, Sharma M, et al. Circulating factor associated with increased glomerular permeability to albumin in recurrent focal segmental glomerulosclerosis. N Engl J Med 1996 ; 334 : 878–883. [Google Scholar]
- Lagrue G, Branellec A, Niaudet P, et al. Transmission of nephrotic syndrome to two neonates. Spontaneous regression. Presse Med 1991 ; 20 : 255–257. [Google Scholar]
- Gallon L, Leventhal J, Skaro A, et al. Resolution of recurrent focal segmental glomerulosclerosis after retransplantation. N Engl J Med 2012 ; 366 : 1648–1649. [Google Scholar]
- Shalhoub RJ. Pathogenesis of lipoid nephrosis: a disorder of T-cell function. Lancet 1974 ; 2 : 556–560. [CrossRef] [PubMed] [Google Scholar]
- Janeway CA. The management of nephrosis. Pediatrics 1948 ; 2 : 705. [PubMed] [Google Scholar]
- Lagrue G, Xheneumont S, Branellec A, et al. A vascular permeability factor elaborated from lymphocytes. I. Demonstration in patients with nephrotic syndrome. Biomedicine 1975 ; 23 : 37–40. [PubMed] [Google Scholar]
- Benz K, Dotsch J, Rascher W, Stachel D. Change of the course of steroid-dependent nephrotic syndrome after rituximab therapy. Pediatr Nephrol 2004 ; 19 : 794–797. [CrossRef] [PubMed] [Google Scholar]
- Yoo TH, Pedigo CE, Guzman J, et al. Sphingomyelinase-like phosphodiesterase 3b expression levels determine podocyte injury phenotypes in glomerular disease. J Am Soc Nephrol 2015 ; 26 : 133–147. [Google Scholar]
- Ollero M, Sahali D. Inhibition of the VEGF signalling pathway and glomerular disorders. Nephrol Dial Transplant 2015 ; 30 : 1449–1455. [CrossRef] [PubMed] [Google Scholar]
- Delville M, Sigdel TK, Wei C, et al. A circulating antibody panel for pretransplant prediction of FSGS recurrence after kidney transplantation. Sci Transl Med 2014; 6 : 256ra136. [CrossRef] [PubMed] [Google Scholar]
- Garin EH, Mu W, Arthur JM, et al. Urinary CD80 is elevated in minimal change disease but not in focal segmental glomerulosclerosis. Kidney Int 2010 ; 78 : 296–302. [CrossRef] [PubMed] [Google Scholar]
- Yu CC, Fornoni A, Weins A, et al. Abatacept in B7-1-positive proteinuric kidney disease. N Engl J Med 2013 ; 369 : 2416–2423. [Google Scholar]
- Lai KW, Wei CL, Tan LK, et al. Overexpression of interleukin-13 induces minimal-change-like nephropathy in rats. J Am Soc Nephrol 2007 ; 18 : 1476–1485. [Google Scholar]
- McCarthy ET, Sharma M, Savin VJ. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 2010 ; 5 : 2115–2121. [CrossRef] [PubMed] [Google Scholar]
- Raveh D, Shemesh O, Ashkenazi YJ, et al. Tumor necrosis factor-alpha blocking agent as a treatment for nephrotic syndrome. Pediatr Nephrol 2004 ; 19 : 1281–1284. [CrossRef] [PubMed] [Google Scholar]
- Bakker WW, Baller JF, van Luijk WH. A kallikrein-like molecule and plasma vasoactivity in minimal change disease. Increased turnover in relapse versus remission. Contrib Nephrol 1988; 67 : 31–6. [CrossRef] [PubMed] [Google Scholar]
- Wei C, Moller CC, Altintas MM, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med 2008 ; 14 : 55–63. [CrossRef] [PubMed] [Google Scholar]
- Kemeny E, Mihatsch MJ, Durmuller U, Gudat F. Podocytes loose their adhesive phenotype in focal segmental glomerulosclerosis. Clin Nephrol 1995 ; 43 : 71–83. [PubMed] [Google Scholar]
- Clement LC, Avila-Casado C, Mace C, et al. Podocyte-secreted angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive nephrotic syndrome. Nat Med 2011 ; 17 : 117–122. [CrossRef] [PubMed] [Google Scholar]
- Sever S, Trachtman H, Wei C, Reiser J. Is there clinical value in measuring suPAR levels in FSGS?. Clin J Am Soc Nephrol 2013 ; 8 : 1273–1275. [CrossRef] [PubMed] [Google Scholar]
- Schwartz MM, Evans J, Bain R, Korbet SM. Focal segmental glomerulosclerosis: prognostic implications of the cellular lesion. J Am Soc Nephrol 1999 ; 10 : 1900–1907. [Google Scholar]
- Campbell KN, Tumlin JA. Protecting podocytes: a key target for therapy of focal segmental glomerulosclerosis. Am J Nephrol 2018 ; 47 : suppl 1 14–29. [CrossRef] [PubMed] [Google Scholar]
- Sharma R, Sharma M, McCarthy ET, et al. Components of normal serum block the focal segmental glomerulosclerosis factor activity in vitro. Kidney Int 2000 ; 58 : 1973–1979. [CrossRef] [PubMed] [Google Scholar]
- Harita Y, Ishizuka K, Tanego A, et al. Decreased glomerular filtration as the primary factor of elevated circulating suPAR levels in focal segmental glomerulosclerosis. Pediatr Nephrol 2014 ; 29 : 1553–1560. [CrossRef] [PubMed] [Google Scholar]
- Faul C, Donnelly M, Merscher-Gomez S, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med 2008 ; 14 : 931–938. [CrossRef] [PubMed] [Google Scholar]
- Beaudreuil S, Zhang X, Florence H, et al. Circulating CASK is associated with recurrent focal segmental glomerulosclerosis after transplantation. PLoS One 2019 (sous presse). [Google Scholar]
- Müller-Deile J, Schenk H, Schroder P, et al. Circulating factors cause proteinuria in parabiotic zebrafish. Kidney Int 2019 Mar 8. pii: S0085–2538(19)30232–7. doi: 10.1016/j.kint.2019.02.013. [Google Scholar]
Liste des figures
|
Figure 1. Cinétique du développement rénal chez l’homme. |
| Dans le texte | |
|
Figure 2. A. Histologie d’un glomérule (Gl) sain chez l’homme (coloration hémalun/éosine x630). Le sang arrive au Gl par l’artériole afférente, il est distribué aux capillaires glomérulaires soutenus par les cellules mésangiales et la matrice qu’elles produisent. Le cytoplasme fenestré des cellules endothéliales des capillaires est orienté vers la membrane basale glomérulaire (MBG) localisée directement à leur contact. Les podocytes reposent sur cette MBG. L’ultrafiltration du sang à travers ces différentes barrières donne naissance à l’urine primitive déversée dans l’espace urinaire et recueillie au pôle tubulaire à l’opposé du pôle vasculaire. L’ultrafiltat sera évacué par le tube contourné proximal. Le sang quitte le Gl par l’artériole efférente. B. Les pédicelles enchevêtrés des podocytes de l’épithélium viscéral de la capsule de Bowman sont en contact avec la MBG et délimitent entre eux l’espace de filtration ou diaphragme de fente. L’autre versant de la MBG est tapissé par l’endothélium du Gl qui limite l’interface sanguine. C. Représentation de quelques molécules qui interviennent au niveau du diaphragme de fente et de la surface basale des podocytes. Le composant majeur du diaphragme est la néphrine. Ces molécules présentent des interactions entre elles et une molécule en forme de harpon: la podocine. Ce complexe moléculaire est lié au cytosquelette d’actine par un complexe intracytoplasmique (non détaillé) déterminé par la protéine associée à CD2 (CD2AP). Le réseau d’actine est régulé en particulier par l’a-actinine-4 mais aussi par la synaptopodine. Au niveau de la surface basale, les molécules d’adhérence telles que l’intégrine a3b1 et l’a-dystroglycane sont liées à la laminine tandis que l’intégrine avb3 est associée à la vitronectine dans la MBG. C’est la sous-unité b des intégrines qui intervient. Ces intégrines sont couplées au réseau d’actine par le complexe taline/vinculine/paxilline tandis que l’a/b-dystroglycane est liée à l’actine par la molécule d’urotrophine. Les podocytes expriment à leur surface des récepteurs d’interleukines et les molécules CD40 et CD80. Ils sont aussi capables d’endocytose. |
| Dans le texte | |
|
Figure 3. Microscopie électronique à balayage d’un rein normal montrant les podocytes avec leurs pédicelles enchevêtrés et les diaphragmes de fente. Comparer avec la Figure 2B. |
| Dans le texte | |
|
Figure 4. A. Microscopie optique d’un rein présentant une HSF où l’on peut voir un glomérule abîmé par la fibrose (coloration au trichrome de Masson x10). B. Microscopie électronique d’une coupe de rein à LGM montrant l’aspect des pédicelles des podocytes. Dans différentes zones, ces pédicelles sont fusionnés. Cette photo montre aussi l’aspect de la MBG et de l’endothélium à cytoplasme fenestré des capillaires glomérulaires. GL : glomérulaire ; MBG : membrane basale glomérulaire. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.