")
")
| Issue |
Med Sci (Paris)
Volume 24, Number 4, Avril 2008
|
|
|---|---|---|
| Page(s) | 407 - 414 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2008244407 | |
| Published online | 15 April 2008 | |
Glycéronéogenèse et PEPCK-C
Cibles pharmacologiques dans le diabète de type 2
Glyceroneogenesis and PEPCK-C: pharmacological targets in type 2 diabetes
Inserm UMR-S 747 ; Université Paris Descartes, Centre universitaire des Saints-Pères, 45, rue des Saints-Pères, 75006 Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Un des liens entre obésité, résistance à l’insuline et diabète de type 2 incrimine les acides gras non estérifiés (AGNE) issus de l’hydrolyse des triglycérides dans le tissu adipeux. Une part importante de ces AGNE est ré-estérifiée immédiatement en triglycérides. La ré-estérification requiert la glycéronéogenèse ou synthèse de glycérol-3-phosphate qui, notamment à jeun, provient de substrats non glucidiques. Plus le recyclage des AGNE est important, moins ils sont libérés. La glycéronéogenèse et l’expression de son enzyme clé, la PEPCK-C, sont sélectivement induites par les thiazolidinediones hypolipidémiantes et antidiabétiques qui réduisent ainsi l’efflux des AGNE, leur taux circulant et l’insulinorésistance.
Abstract
Obesity is a major risk factor for insulin resistance and type 2 diabetes. The link between hypertrophied adipose tissue and this pathology is thought to be non-esterified fatty acids (NEFA) arising from adipocyte lipolysis. Sustained increase in plasma NEFA induces insulin resistance. In adipocytes, a significant part of lipolytic NEFA is re-esterified to triacylglycerol. Reesterification requires glycerol-3-phosphate which, during fasting, is synthesized from lactate, pyruvate or certain amino acids in a metabolic pathway named glyceroneogenesis. The key enzyme in this pathway is the cytosolic phosphoenolpyruvate carboxykinase (PEPCK-C). In this review, we postulate that thiazolidinediones exert their hypolipidemic and antidiabetic effects in adipose tissue at least in part through a rapid and selective induction of PEPCK-C gene transcription leading to increased PEPCK-C and glyceroneogenesis. Subsequent fatty acid re-esterification participates in the reduction in blood NEFA and insulin resistance.
© 2008 médecine/sciences - Inserm / SRMS
Obésité, acides gras circulants et insulinorésistance
L’insulinorésistance est une réaction diminuée de l’organisme en réponse à l’insuline, conduisant in fine à une moindre utilisation du glucose, donc à l’hyperglycémie et à l’installation du diabète de type 2.
Il existe des prédispositions génétiques au développement de l’insulino-résistance mais des facteurs de risque aggravent sa prévalence. Ainsi, un déséquilibre entre apport calorique et dépense énergétique entraîne une augmentation de la masse adipeuse, principal facteur de risque de cette pathologie. L’obésité qui en résulte est souvent associée à l’insulinorésistance et au diabète de type 2 [1]. Un article de la revue Science publié en 1990 par Denis McGarry a pour titre « What if Minkowski had been ageusic ? an alternative angle on diabetes » (que ce serait-il passé si Minkowski 1 n’avait pas eu le sens du goût ? une vue alternative sur le diabète) [2]. Dans ce cas, Minkowski n’aurait pas pu goûter le sucre des urines mais en revanche il aurait pu sentir l’acétone présente dans celles-ci et en conclure que le diabète est une maladie des lipides, plus particulièrement des acides gras pourvoyeurs de corps cétoniques retrouvés dans les urines lors de cette pathologie. Dans cette hypothèse les acides gras non estérifiés (AGNE), notamment ceux libérés par le tissu adipeux présent en excès chez l’obèse, seraient la clé du problème métabolique lié au diabète de type 2 (Figure 1). L’hypertrophie des adipocytes conduirait à un efflux excessif d’AGNE vers le foie et les muscles squelettiques entraînant un phénomène de compétition de substrat avec l’utilisation du glucose, selon le postulat du cycle de Randle2 [3, 35]. Par ailleurs, le stockage ectopique de ces AGNE sous forme de triglycérides conduirait à l’accumulation d’intermédiaires métaboliques, comme les acyl-CoA, les diacylglycérols et les céramides, qui seraient à l’origine de la moindre utilisation de glucose par le muscle, par leur interférence avec la cascade de signalisation associée à l’insuline (Figure 2). Cette interférence s’exercerait en favorisant des phosphorylations inhibitrices sur sérine ou thréonine du substrat du récepteur de l’insuline (IRS), bloquant ainsi le recrutement des protéines telles que la PI3 kinase et les processus qui impliquent cette enzyme (translocation de Glut4 à la membrane dans le muscle notamment) [4] (Figure 2). Ainsi, le glucose serait beaucoup moins utilisé par les tissus sensibles à l’insuline, ce qui aurait pour conséquence de favoriser l’hyperglycémie. À long terme, l’augmentation persistante des AGNE plasmatiques conduirait à la destruction des cellules β du pancréas productrices d’insuline, ce qui rendrait les diabétiques insulino-« requérants ».
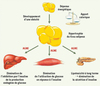 |
Figure 1. Rôle central du tissu adipeux et des acides gras dans l’insulino-résistance. Lors d’un déséquilibre de la balance énergétique, l’hypertrophie du tissu adipeux entraîne une libération excessive d’acides gras non-estérifiés (AGNE) dans le flux sanguin. Ces AGNE peuvent alors induire une insulinorésistance et avoir des effets délétères sur différents organes tels que le foie ou le muscle. À plus long terme, un phénomène de lipotoxicité peut apparaître au niveau du pancréas et altérer la sécrétion d’insuline. |
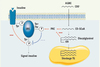 |
Figure 2. Altération de la cascade de signalisation de l’insuline par les acides gras non estérifiés. L’excès d’AGNE circulants se traduit par un stockage ectopique sous forme de triglycérides (TG) dans des organes comme le foie ou le muscle. Cela conduit à l’accumulation d’intermédiaires métaboliques acyl-CoA ou diacylglycérol, qui favorisent, via la protéine kinase C (PKC), des phosphorylations sur sérine ou thréonine du substrat du récepteur de l’insuline (IRS) qui, au contraire des phosphorylations sur tyrosine, empêchent le recrutement de la PI3 kinase, bloquant ainsi le signal porté par l’insuline. |
Une telle élévation des AGNE pourrait provenir d’un défaut de leur stockage dans le tissu adipeux à la suite d’un repas, d’une altération de leur oxydation hépatique ou musculaire ou encore de leur libération excessive par le tissu adipeux notamment lors du jeûne. Dans cette synthèse nous focalisons notre argumentation sur ce dernier point, en postulant que les mécanismes contrôlant la libération des AGNE par le tissu adipeux constitueraient des cibles intéressantes pour une stratégie thérapeutique anti-diabétique.
Pas de guerre de TROIS pour les acides gras
Les ouvrages de biochimie décrivent la libération des AGNE du tissu adipeux comme issue du processus de lipolyse des triglycérides de stockage par l’action de la lipase hormono-sensible (LHS). En réalité ce schéma est très incomplet et ne permet pas d’expliquer le phénomène de lipolyse dans sa globalité. D’une part, alors que la LHS hydrolyse les triglycérides et les diglycérides, elle ne reconnaît pas les monoglycérides. Une monoglycéride lipase (MGL) hydrolyse ces derniers. D’autre part, l’utilisation de souris transgéniques a démontré que la LHS ne pouvait être la seule hydrolase des triglycérides mise en action, observation confortée par la mise en évidence récente d’une autre enzyme du tissu adipeux, la lipase des triglycérides (ATGL), qui hydrolyse spécifiquement ceux-ci [5]. Il est donc désormais à peu près admis que ATGL, LHS et MGL agissent séquentiellement pour aboutir à l’hydrolyse complète des triglycérides. Enfin, on a identifié une classe de protéines appelées périlipine, adipophiline et TIP47, nécessaires à l’ancrage de la LHS à la vacuole lipidique ce qui lui facilite l’accession au substrat [6, 7].
Toutefois, même si ce schéma métabolique commence à être plus élaboré, une observation cruciale n’est pratiquement jamais prise en considération pour décrire la biodisponibilité réelle des AGNE, c’est-à-dire la mesure de leur quantité délivrée par l’adipocyte dans le sang en comparaison de celle du glycérol. En effet, selon la stœchiométrie, il faut s’attendre à ce que trois molécules d’AGNE et une molécule de glycérol soient libérées dans le sang à partir d’une molécule de triglycéride de stockage. Or, l’expérience prouve que ce n’est pratiquement jamais le cas [8]. Même si la proportion relative des AGNE et du glycérol libérés dans le milieu d’incubation d’adipocytes ou dans le sang varie selon les conditions expérimentales et l’espèce considérée, le rapport AGNE/glycérol est toujours inférieur à trois [9, 10].
De fait, le flux lipolytique est en permanence contrebalancé par la ré-estérification en triglycérides d’une partie des AGNE issus de la lipolyse. Cette ré-estérification, qui permet une régulation fine de la sortie des AGNE sans affecter celle du glycérol, requiert la synthèse de glycérol-3-phosphate (G3P) (Figure 3). Ce dernier, en période de forte activité lipolytique, ne peut provenir du glucose dont le transport et le métabolisme sont fortement réduits lors du jeûne. Par ailleurs, la faible activité glycérol kinase présente dans le tissu adipeux blanc ne permet pas d’expliquer les taux élevés de ré-estérification d’AGNE, à partir du glycérol issu de la lipolyse. Il y a une quarantaine d’années, Richard Hanson (Cleveland, OH, États-Unis) et ses collègues ont démontré que le tissu adipeux est capable d’utiliser le pyruvate et le lactate pour synthétiser de novo le G3P via une voie métabolique appelée glycéronéogenèse [11]. Cette voie métabolique pourrait ainsi permettre non seulement la ré-estérification des AGNE en période de lipolyse mais aussi participer à l’estérification, dans le tissu adipeux, des AGNE provenant des lipoprotéines circulantes lorsque la glycolyse est réduite, c’est-à-dire à distance du pic d’insuline, ou lors d’un régime riche en lipides et dépourvu de glucides [12]. Comment fonctionne cette voie métabolique méconnue bien qu’ayant un rôle crucial dans la biodisponibilité des AGNE ?
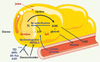 |
Figure 3. Métabolisme adipocytaire des acides gras nonestérifiés en période de jeûne. Au cours du jeûne, les réserves en triglycérides sont mobilisées afin de subvenir aux besoins énergétiques de l’organisme. La lipolyse, responsable de la formation d’AGNE est en permanence contrebalancée par la ré-estérification d’une partie de ces AGNE. Celle-ci requiert la synthèse de glycérol-3-phosphate (G3P) qui provient principalement du lactate ou du pyruvate grâce à une voie appelée glycéronéogenèse et dont l’enzyme clé est la PEPCK-C. En effet, les autres voies de production du G3P sont quantitativement négligeables car la glycérol kinase (GyK) est faiblement exprimée dans les adipocytes, et la glycolyse est réduite lors du jeûne ou plus généralement lorsque le niveau circulant d’insuline est bas. Par sa fonction de synthèse de G3P, permettant la ré-estérification des AGNE, la glycéronéogenèse contrôle le flux sortant de ces AGNE dans la circulation sanguine. Les TZD (TZD) hypolipidémiantes et antidiabétiques, l’AMPc, l’AR-9cis et les acides gras polyinsaturés (AGPI) sont des inducteurs du gène PEPCK-C, conduisant à l’augmentation de la protéine PEPCK-C et de la glycéronéogenèse. Les glucocorticoïdes répriment toutes les stimulations connues de la PEPCK-C dans le tissu adipeux, ce qui entraîne une augmentation de l’efflux d’AGNE. |
Imprononçables mais tellement utiles : glycéronéogenèse et phosphoénolpyruvate carboxykinase
L’enzyme clé de la glycéronéogenèse est l’isoforme cytosolique de la phosphoénolpyruvate carboxykinase3 (PEPCK-C ; EC 4.1.1.32) (Figure 3) [11, 34]. Cette enzyme catalyse la décarboxylation oxydative de l’oxaloacétate en phosphoénolpyruvate et nécessite du GTP pour cette réaction. Elle est surtout connue comme étant l’enzyme majeure de la gluconéogenèse hépatique et rénale mais elle s’exprime aussi très fortement dans trois autres tissus qui ne produisent pas de glucose, le tissu adipeux, l’intestin et la glande mammaire lors de la lactation. La glycéronéogenèse est donc considérée comme une version abrégée de la gluconéogenèse. Les mesures de l’activité enzymatique PEPCK-C ont montré que celle-ci reflète parfaitement le flux glycéronéogénique. Pour cette raison, les études se sont focalisées sur la régulation de l’expression de la PEPCK-C.
Une proportion importante des enzymes du métabolisme intermédiaire est régulée soit allostériquement soit par des modifications post-traductionnelles : phosphorylation ou glycosylation. Ce n’est pas le cas de la PEPCK-C dont il n’existe aucune régulation post-traductionnelle connue à ce jour. Ainsi, toute modification de son activité enzymatique est due à une variation de la quantité d’enzyme, elle même liée à une modulation de la balance synthèse/dégradation de son ARNm. La régulation de la transcription de ce gène, donc du promoteur PEPCK-C, a été étudiée de façon approfondie. Parmi les nombreux éléments de réponse aux hormones identifiés dans le promoteur du gène PEPCK-C de rat [13], deux éléments pouvant fixer l’hétérodimère des récepteurs nucléaires PPARγ et RXR ont été mis en évidence par Tontonoz et al. [14]. Ces 2 séquences d’ADN, de type « direct repeat 1 » (DR1 - AGGTCAnAGGTCA), sont dites de réponse aux proliférateurs de peroxysomes (PPRE) (Figure 4). Elles sont conservées chez l’homme et les rongeurs et sont localisées respectivement à environ 0,4 (PCK1) et 1 (PCK2) kilobases en amont du site de démarrage de la transcription du gène (Figure 4). Elmus Beale (Lubbock, TX, États-Unis) et ses collaborateurs ont clairement montré que PCK2 est un élément particulièrement important car indispensable à une forte expression du gène dans le tissu adipeux, sans affecter celle du foie [15].
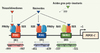 |
Figure 4. Les éléments de réponse du promoteur du gène PEPCK-C de rat impliqués dans la réponse aux thiazolidinediones et aux réxinoïdes. Les TZD, agonistes du récepteur PPARγ, activent la transcription du gène PEPCK-C via l’élément de réponse PCK2 localisé dans une région du promoteur qui confère la spécificité d’expression du gène dans les adipocytes, à environ 1kb en amont du site de démarrage de la transcription (+1). PCK1 est aussi capable de lier l’hétérodimère PPARγ/RXR in vitro mais ne répond pas aux TZD. Les réxinoïdes, ligands de RXR, activent le gène via 3 éléments de réponse agissant de concert : PCK2 et PCK1 et RARE2. Alors que PCK2 et PCK1 sont des direct repeat 1 (DR1) en orientation inverse, RARE2 est un DR5 capable de fixer l’hétérodimère RXR/RAR. Les acides gras polyinsaturés sont eux aussi des activateurs du gène PEPCK-C, cependant, les éléments de réponse du promoteur responsables de cette activation ne sont, à ce jour, pas connus. Il semble cependant que PPARγ ne soit pas impliqué dans l’effet des acides gras [30]. |
Pourquoi refaire du gras n’est pas si mauvais : le cas des thiazolidinediones
Parmi les molécules anti-diabétiques disponibles en clinique, la classe des thiazolidinediones (TZD), dont la rosiglitazone (Avandia®) est un prototype, appartient à la famille des insulino-sensibilisateurs. Leur mécanisme d’action n’est pas complètement élucidé mais il est cependant admis qu’il fait intervenir PPARγ, puisque les TZD sont des agonistes très sélectifs de ce récepteur [16]. Ainsi, ces molécules exerceraient leur action précoce en régulant des gènes cibles de PPARγ dans le tissu adipeux puisque c’est dans ce tissu que l’on retrouve les plus forts taux d’expression de ce récepteur. Des études in vitro ont montré que les TZD sont adipogéniques, c’est-à-dire qu’elles favorisent la formation de nouveaux adipocytes à partir de leurs précurseurs, les préadipocytes ou même d’autres précurseurs dérivant de la moelle osseuse [17, 18]. Ces « jeunes » adipocytes seraient insulino-sensibles, contrairement aux « vieux et gros » adipocytes qui le seraient moins. Ils seraient donc capables de métaboliser plus efficacement le glucose et les AGNE, ce qui favoriserait le retour à une glycémie normale. Or, très rapidement, ces nouveaux adipocytes devraient grossir et perdre leur sensibilité à l’insuline, annulant de ce fait les effets bénéfiques des TZD. Parmi les mécanismes d’action supposés des TZD, des modifications du profil de sécrétion d’adipokines, notamment de l’adiponectine, ont aussi été évoquées [19]. Nos résultats nous ont conduit à élaborer une autre théorie qui fait intervenir le métabolisme des AGNE, plus précisément leur recyclage.
La glycéronéogenèse du tissu adipeux humain, comme celle des rongeurs, est fortement induite par les TZD qui ont pour cible primaire le gène PEPCK-C [9, 10, 20–23]. L’induction de cette enzyme entraîne une augmentation de la ré-estérification des AGNE conduisant à une diminution rapide et conséquente de leur libération par le tissu adipeux. L’inhibition spécifique de l’activité enzymatique PEPCK-C par le mercaptopicolinate prévient complètement ces effets démontrant ainsi que cette enzyme joue le rôle principal dans ce processus [9]. La PEPCK-C est donc un acteur crucial dans l’effet hypolipidémiant précoce des TZD qui semble être primordial pour leurs propriétés insulino-sensibilisatrices. Ce mécanisme est intéressant mais n’est compréhensible qu’à trois conditions. D’une part, puisque les TZD font « maigrir moins vite » les adipocytes, un mécanisme de dépense énergétique doit se mettre en place. En cela, la démonstration récente que ces molécules augmentent la capacité de ß-oxydation des acides gras est convaincante [24]. D’autre part, il faut postuler une spécificité d’action des TZD sur les adipocytes, car la PEPCK-C des hépatocytes est gluconéogénique et son induction entraînerait une augmentation de la production de glucose, ce qui aurait des conséquences délétères. Enfin, seul un nombre restreint de gènes devrait être affecté.
Quand spécificité rime avec efficacité
Nos résultats montrent que les TZD induisent le gène PEPCK-C dans les adipocytes mais pas dans les hépatocytes ou les cellules d’hépatomes, peut-être simplement parce que PPARγ n’y est pas exprimé [20, 25]. Cette spécificité cellulaire permet leur action antidiabétique. Par ailleurs, l’expression de ce gène et la glycéronéogenèse du tissu adipeux de rat sont plus élevées et répondent mieux aux TZD dans le dépot viscéral que sous-cutané [9]. Cette observation est à mettre en parallèle du fait que le développement excessif de la masse adipeuse viscérale est plus propice au développement d’un état de résistance à l’insuline.
Les agonistes de PPARγ sont des activateurs transcriptionnels directs, très rapides et puissants de PEPCK-C dans les adipocytes. Parmi toute une série d’autres gènes décrits comme régulés par les TZD, PEPCK-C a une place à part, répondant dans les minutes qui suivent l’ajout de l’agent sur les cellules, un effet qui persiste à long terme (au moins trois mois chez l’homme) [23]. Cette sélectivité génique d’action des agonistes PPARγ met en exergue l’importance de ce gène. La littérature foisonne d’exemples de « gènes régulés par les TZD » mais très peu ont été décrits expérimentalement comme des cibles primaires - à savoir rapides et directes - de cette classe de médicaments. Le cas du gène codant la glycérol kinase est exemplaire à ce sujet car s’il est décrit comme induit par les TZD, une controverse est née à ce propos, peut-être tout simplement parce qu’il est influencé indirectement, et donc tardivement, par ces molécules [26, 27]. Alors que les séquences cibles potentielles de PPARγ - PCK1 et PCK2 - sont présentes dans le promoteur du gène PEPCK-C, l’effet de la rosiglitazone ne fait intervenir que PCK2, ce qui signifie que toute séquence de type PPRE présente dans le promoteur d’un gène n’est pas nécessairement fonctionnelle (Figure 4). Par ailleurs, PPARγ agit sous la forme d’un hétérodimère permissif avec RXR, les deux pouvant être activés par leurs agonistes respectifs. Nos travaux en cours montrent effectivement que les agonistes de RXR (rexinoïdes) activent eux-aussi la glycéronéogenèse et induisent la transcription du gène PEPCK-C.
De la pharmacologie à la physiologie : effet des β-agonistes, des acides gras et des glucocorticoïdes
Les agonistes β-adrénergiques, via la production d’AMPc et les acides gras insaturés, plus particulièrement les polyinsaturés oméga-3, sont des activateurs transcriptionnels directs puissants du gène PEPCK-C dans les adipocytes [12, 28, 29]. Le mécanisme moléculaire de l’activation par les oméga-3 semble différer de celui des TZD même si, comme ces dernières, les acides gras agissent de façon directe et sélective sur ce gène [30]. Si, comme nous nous y attendons, les oméga-3 réduisent aussi le niveau circulant des AGNE issus de la lipolyse, ce mécanisme pourrait participer à l’action bénéfique de cette classe d’AGNE sur l’insulino-résistance. À l’inverse, les glucocorticoïdes sont des inhibiteurs de tous les inducteurs du gène PEPCK-C dans les adipocytes. Le mécanisme de cette action inhibitrice est partiellement connu et diffère de celui qui a été abondamment décrit dans le foie où les glucocorticoïdes sont des activateurs transcriptionnels de ce même gène. L’action opposée des glucocorticoïdes dans ces deux tissus a un sens physiologique puisque ce sont des hormones de stress qui conduisent à un apport d’énergie dans le sang - production de glucose par le foie et réduction du frein à la libération des AGNE par le tissu adipeux -. Les glucocorticoïdes participent donc à la mobilisation accrue des AGNE en période de stress.
Quelle implication dans l’obésité et le diabète de type 2 ?
La glycéronéogenèse de base du tissu adipeux diminue lorsque l’index de masse corporelle augmente [10]. Il existe donc une altération de la capacité des « gros » adipocytes à ré-estérifier les AGNE, ce qui est en accord avec la libération accrue de ces derniers. En revanche, l’effet stimulateur de la rosiglitazone persiste. Par ailleurs, dans différentes cohortes de patients diabétiques de type 2, le traitement avec une thiazolidinedione entraîne une augmentation de l’expression du gène PEPCK-C dans le tissu adipeux, sans affecter l’expression de toute une série d’autres gènes potentiellement sensibles à cette série de molécules, dont notamment celui codant la glycérol kinase [23, 31]. Dans un modèle de rat insulino-résistants, la rosiglitazone administrée par voie orale induit une augmentation de l’ARNm PEPCK-C et de l’activité de l’enzyme dans différents dépôts de tissu adipeux sous-cutanés et profonds. Cela s’accompagne d’une moindre libération d’AGNE par le tissu adipeux de ces rats et, en conséquence, d’une réduction de leur concentration plasmatique ainsi que de celle des triglycérides [23]. Par conséquent, la glycéronéogenèse et l’induction de son gène clé apparaissent diminuées et/ou dérégulées chez les obèses diabétiques.
Il y a plusieurs années, une liaison génétique entre la région du gène PEPCK-C sur le chromosome 20q13 humain et le diabète de type 2 a été mise en évidence [32]. Par la suite, un polymorphisme de nucléotides simples (SNP) associé à ce type de diabète a été localisé dans un élément putatif de réponse à l’AMPc du promoteur du gène [33]. Il faut noter que la transcription du gène PEPCK-C est stimulée par l’AMPc dans le foie comme dans le tissu adipeux. Il faut s’attendre à ce qu’un tel polymorphisme affecte potentiellement l’expression du gène dans les deux tissus. Nous nous sommes intéressés à la région d’ADN englobant l’élément PCK2, qui confère la sélectivité d’expression du gène dans le tissu adipeux (voir ci-dessus). Nous avons identifié, de part et d’autre de PCK2, deux SNP en fort déséquilibre de liaison et associées à la sévérité de certains paramètres glycémiques du diabétique de type 2 [21]. La détermination d’un lien de causalité entre ces SNP et la pathologie nécessitera l’analyse fonctionnelle du promoteur portant ces séquences polymorphes.
Conclusions
L’activation rapide de la glycéronéogenèse adipocytaire par des molécules hypolipidémiantes et antidiabétiques participe à la séquestration des lipides dans les adipocytes qui sont les seules cellules de l’organisme spécialisées dans le stockage de triglycérides. Cela a pour conséquence la réduction du flux d’AGNE vers les organes tels que le foie, le muscle ou le pancréas, diminuant ainsi l’insulino-résistance. L’effet hypolipidémiant précoce de ces molécules, auquel contribue l’induction de la PEPCK-C et de la glycéronéogenèse du tissu adipeux, semble être essentiel pour expliquer leur effet antidiabétique.
Le lien entre pancréas et diabète sucré n’est établi expérimentalement qu’en 1889, lorsque les médecins allemands, Oskar Minkowski (1858-1931) et Josef Von Mering (1849-1908), montrent que l’ablation du pancréas chez un chien provoque le diabète sucré.
Il y a une quarantaine d’années, Sir Philip Randle et ses collaborateurs ont démontré, à partir d’expériences réalisées in vitro sur le coeur de rat isolé et perfusé, l’existence d’un cycle « glucose-acides gras ». Dans ces expériences, une augmentation de la concentration des AGL dans le milieu de perfusion conduisait à une réduction du captage et de l’utilisation du glucose par le myocarde. Ils avaient ensuite généralisé ces observations au muscle squelettique, et suggéré qu’un excès d’AGL pourrait in vivo contribuer à la diminution de l’utilisation du glucose et au développement du diabète de type 2 [35].
Cette enzyme est la même que celle qui, surexprimée dans les muscles de souris transgéniques rend ces souris « marathoniennes » [34].
Références
- Keller U. From obesity to diabetes. Int J Vitam Nutr Res 2006; 76 : 172–7. [Google Scholar]
- McGarry J. D. What if Minkowski had been ageusic ? An alternative angle on diabetes. Science 1992; 258 : 766–70. [Google Scholar]
- Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963; 1 : 785–9. [Google Scholar]
- Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest 2000; 106 : 171–6. [Google Scholar]
- Zimmermann R, Strauss JG, Haemmerle G, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004; 306 : 1383–6. [Google Scholar]
- Londos C, Sztalryd C, Tansey JT, Kimmel AR. Role of PAT proteins in lipid metabolism. Biochimie 2005; 87 : 45–9. [Google Scholar]
- Tai ES, Ordovas JM. The role of perilipin in human obesity and insulin resistance. Curr Opin Lipidol 2007; 18 : 152–6. [Google Scholar]
- Vaughan M. The production and release of glycerol by adipose tissue incubated in vitro. J Biol Chem 1962; 237 : 3354–8. [Google Scholar]
- Tordjman J, Chauvet G, Quette J, et al. Thiazolidinediones block fatty acid release by inducing glyceroneogenesis in fat cells. J Biol Chem 2003; 278 : 18785–90. [Google Scholar]
- Leroyer SN, Tordjman J, Chauvet G, et al. Rosiglitazone controls fatty acid cycling in human adipose tissue by means of glyceroneogenesis and glycerol phosphorylation. J Biol Chem 2006; 281 : 13141–9. [Google Scholar]
- Ballard FJ, Hanson RW, Leveille GA. Phosphoenolpyruvate carboxykinase and the synthesis of glyceride-glycerol from pyruvate in adipose tissue. J Biol Chem 1967; 242 : 2746–50. [Google Scholar]
- Antras-Ferry J, Robin P, Robin D, Forest C. Fatty acids and fibrates are potent inducers of transcription of the phosphenolpyruvate carboxykinase gene in adipocytes. Eur J Biochem 1995; 234 : 390–6. [Google Scholar]
- Chakravarty K, Cassuto H, Reshef L, Hanson RW. Factors that control the tissue-specific transcription of the gene for phosphoenolpyruvate carboxykinase-C. Crit Rev Biochem Mol Biol 2005; 40 : 129–54. [Google Scholar]
- Tontonoz P, Hu E, Devine J, et al. PPAR gamma 2 regulates adipose expression of the phosphoenolpyruvate carboxykinase gene. Mol Cell Biol 1995; 15 : 351–7. [Google Scholar]
- Devine JH, Eubank DW, Clouthier DE, et al. Adipose expression of the phosphoenolpyruvate carboxykinase promoter requires peroxisome proliferator-activated receptor gamma and 9-cis-retinoic acid receptor binding to an adipocyte-specific enhancer in vivo. J Biol Chem 1999; 274 : 13604–12. [Google Scholar]
- Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs : from orphan receptors to drug discovery. J Med Chem 2000; 43 : 527–50. [Google Scholar]
- Okuno A, Tamemoto H, Tobe K, et al. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese Zucker rats. J Clin Invest 1998; 101 : 1354–61. [Google Scholar]
- Crossno JT Jr, Majka SM, Grazia T, et al. Rosiglitazone promotes development of a novel adipocyte population from bone marrow-derived circulating progenitor cells. J Clin Invest 2006; 116 : 3220–8. [Google Scholar]
- Guerre-Millo M. Adipose tissue and adipokines : for better or worse. Diabetes Metab 2004; 30 : 13–9. [Google Scholar]
- Glorian M, Duplus E, Beale EG, et al. A single element in the phosphoenolpyruvate carboxykinase gene mediates thiazolidinedione action specifically in adipocytes. Biochimie 2001; 83 : 933–43. [Google Scholar]
- Duplus E, Benelli C, Reis AF, et al. Expression of phosphoenolpyruvate carboxykinase gene in human adipose tissue : induction by rosiglitazone and genetic analyses of the adipocyte-specific region of the promoter in type 2 diabetes. Biochimie 2003; 85 : 1257–64. [Google Scholar]
- Hallakou S, Doare L, Foufelle F, et al. Pioglitazone induces in vivo adipocyte differentiation in the obese Zucker fa/fa rat. Diabetes 1997; 46 : 1393–9. [Google Scholar]
- Cadoudal T, Blouin JM, Collinet M, et al. Acute and selective regulation of glyceroneogenesis and cytosolic phosphoenolpyruvate carboxykinase in adipose tissue by thiazolidinediones in type 2 diabetes. Diabetologia 2007; 50 : 666–75. [Google Scholar]
- Boden G, Homko C, Mozzoli M, et al. Thiazolidinediones upregulate fatty acid uptake and oxidation in adipose tissue of diabetic patients. Diabetes 2005; 54 : 880–5. [Google Scholar]
- Davies GF, Khandelwal RL, Wu L, et al. Inhibition of phosphoenolpyruvate carboxykinase (PEPCK) gene expression by troglitazone : a peroxisome proliferator-activated receptor-gamma (PPARgamma)-independent, antioxidant-related mechanism. Biochem Pharmacol 2001; 62 : 1071–9. [Google Scholar]
- Guan HP, Li Y, Jensen MV, et al. A futile metabolic cycle activated in adipocytes by antidiabetic agents. Nat Med 2002; 8 : 1122–8. [Google Scholar]
- Tan GD, Debard C, Tiraby C, et al. A “futile cycle” induced by thiazolidinediones in human adipose tissue ? Nat Med 2003; 9 : 811–2. [Google Scholar]
- Franckhauser S, Antras-Ferry J, Robin P, et al. Expression of the phosphoenolpyruvate carboxykinase gene in 3T3-F442A adipose cells : opposite effects of dexamethasone and isoprenaline on transcription. Biochem J 1995; 305 : 65–71. [Google Scholar]
- Duplus E, Glorian M, Tordjman J, et al. Evidence for selective induction of phosphoenolpyruvate carboxykinase gene expression by unsaturated and nonmetabolized fatty acids in adipocytes. J Cell Biochem 2002; 85 : 651–61. [Google Scholar]
- Duplus E, Forest C. Is there a single mechanism for fatty acid regulation of gene transcription ? Biochem Pharmacol 2002; 64 : 893–901. [Google Scholar]
- Bogacka I, Xie H, Bray GA, Smith SR. The effect of pioglitazone on peroxisome proliferator-activated receptor-gamma target genes related to lipid storage in vivo. Diabetes Care 2004; 27 : 1660–7. [Google Scholar]
- El Hani H, Zouali H, Philippi A, et al. Indication for genetic linkage of the phosphoenolpyruvate carboxykinase (PCK1) gene region on chromosome 20q to non-insulin-dependent diabetes mellitus. Diabetes Metab 1996; 22 : 451–4. [Google Scholar]
- Cao H, Van der Veer E, Ban BR, et al. Promoter polymorphism in PCK1 (phosphoenolpyruvate carboxykinase gene) associated with type 2 diabetes mellitus. J Clin Endocrinol Metab 2004; 89 : 898–903. [Google Scholar]
- Hakimi P, Yang J, Casadesus G, et al. Overexpression of the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) in skeletal muscle repatterns energy metabolism in the mouse. J Biol Chem 2007; 282 : 328–44. [Google Scholar]
- Girard J. Rôle des acides gras libres dans la sécrétion et l’action de l’insuline : mécanismes de la lipotoxicité. Med Sci (Paris) 2003; 19 : 827–33. [Google Scholar]
Liste des figures
|
Figure 1. Rôle central du tissu adipeux et des acides gras dans l’insulino-résistance. Lors d’un déséquilibre de la balance énergétique, l’hypertrophie du tissu adipeux entraîne une libération excessive d’acides gras non-estérifiés (AGNE) dans le flux sanguin. Ces AGNE peuvent alors induire une insulinorésistance et avoir des effets délétères sur différents organes tels que le foie ou le muscle. À plus long terme, un phénomène de lipotoxicité peut apparaître au niveau du pancréas et altérer la sécrétion d’insuline. |
| Dans le texte | |
|
Figure 2. Altération de la cascade de signalisation de l’insuline par les acides gras non estérifiés. L’excès d’AGNE circulants se traduit par un stockage ectopique sous forme de triglycérides (TG) dans des organes comme le foie ou le muscle. Cela conduit à l’accumulation d’intermédiaires métaboliques acyl-CoA ou diacylglycérol, qui favorisent, via la protéine kinase C (PKC), des phosphorylations sur sérine ou thréonine du substrat du récepteur de l’insuline (IRS) qui, au contraire des phosphorylations sur tyrosine, empêchent le recrutement de la PI3 kinase, bloquant ainsi le signal porté par l’insuline. |
| Dans le texte | |
|
Figure 3. Métabolisme adipocytaire des acides gras nonestérifiés en période de jeûne. Au cours du jeûne, les réserves en triglycérides sont mobilisées afin de subvenir aux besoins énergétiques de l’organisme. La lipolyse, responsable de la formation d’AGNE est en permanence contrebalancée par la ré-estérification d’une partie de ces AGNE. Celle-ci requiert la synthèse de glycérol-3-phosphate (G3P) qui provient principalement du lactate ou du pyruvate grâce à une voie appelée glycéronéogenèse et dont l’enzyme clé est la PEPCK-C. En effet, les autres voies de production du G3P sont quantitativement négligeables car la glycérol kinase (GyK) est faiblement exprimée dans les adipocytes, et la glycolyse est réduite lors du jeûne ou plus généralement lorsque le niveau circulant d’insuline est bas. Par sa fonction de synthèse de G3P, permettant la ré-estérification des AGNE, la glycéronéogenèse contrôle le flux sortant de ces AGNE dans la circulation sanguine. Les TZD (TZD) hypolipidémiantes et antidiabétiques, l’AMPc, l’AR-9cis et les acides gras polyinsaturés (AGPI) sont des inducteurs du gène PEPCK-C, conduisant à l’augmentation de la protéine PEPCK-C et de la glycéronéogenèse. Les glucocorticoïdes répriment toutes les stimulations connues de la PEPCK-C dans le tissu adipeux, ce qui entraîne une augmentation de l’efflux d’AGNE. |
| Dans le texte | |
|
Figure 4. Les éléments de réponse du promoteur du gène PEPCK-C de rat impliqués dans la réponse aux thiazolidinediones et aux réxinoïdes. Les TZD, agonistes du récepteur PPARγ, activent la transcription du gène PEPCK-C via l’élément de réponse PCK2 localisé dans une région du promoteur qui confère la spécificité d’expression du gène dans les adipocytes, à environ 1kb en amont du site de démarrage de la transcription (+1). PCK1 est aussi capable de lier l’hétérodimère PPARγ/RXR in vitro mais ne répond pas aux TZD. Les réxinoïdes, ligands de RXR, activent le gène via 3 éléments de réponse agissant de concert : PCK2 et PCK1 et RARE2. Alors que PCK2 et PCK1 sont des direct repeat 1 (DR1) en orientation inverse, RARE2 est un DR5 capable de fixer l’hétérodimère RXR/RAR. Les acides gras polyinsaturés sont eux aussi des activateurs du gène PEPCK-C, cependant, les éléments de réponse du promoteur responsables de cette activation ne sont, à ce jour, pas connus. Il semble cependant que PPARγ ne soit pas impliqué dans l’effet des acides gras [30]. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.