")
")
| Issue |
Med Sci (Paris)
Volume 38, Number 8-9, Août-Septembre 2022
|
|
|---|---|---|
| Page(s) | 655 - 662 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2022103 | |
| Published online | 12 September 2022 | |
Pseudo-hypoparathyroïdie et ses variants
Un succès de la médecine translationnelle
Pseudohypoparathyroidism and variants: A translational medicine success story
1
Université Caen-Normandie, UFR santé, 2 rue des Rochambelles, 14032 Caen Cedex 5, France
2
Service de génétique, Centre de référence normand des maladies rares du métabolisme du calcium et du phosphore, CHU de Caen - hôpital Clémenceau, avenue Georges Clémenceau, 14033 Caen, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les pseudohypoparathyroïdies (PHP) sont des maladies rares, caractérisées par une résistance à l’action rénale de la parathormone. Le défaut génétique est localisé au locus GNAS, qui code la sous-unité alpha stimulatrice des protéines G (Gαs). Ce locus est le siège de régulations complexes, épissage alternatif et empreinte parentale éteigant de façon tissu-spécifique l’expression de l’allèle paternel. Des mutations hétérozygotes perte de fonction, des épimutations responsables d’une perte d’expression sont associées à un large spectre pathologique : PHP1A, PHP1B, ossification hétérotopique, ostéodystophie, obésité, retard de croissance in utero, etc., dont les mécanismes restent encore incomplètement connus.
Abstract
Pseudohypoparathyroidism (PHP) is an uncommon disorder which is characterized by end-organ PTH resistance. The genetic defect is located at the GNAS locus that encodes the alpha-subunit of the stimulatory G protein (Gαs) and several splice variants thereof. This complex locus undergoes parental specific methylation changes that result in tissue-specific silencing of the paternal allele. Heterozygous inactivating mutations that disrupt Gαs function or epigenetics changes that impair Gαs expression contribute to a wide clinical spectrum of the disease: PHP1A, PHP1B, osseous heteroplasia, osteodystrophy, obesity, intrauterine growth retardation… whose mechanisms at the molecular level remain unresolved.
© 2022 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (© DR).
Quatre-vingts ans de recherches clinique, biologique et génétique ont permis de décrypter les bases physiopathologiques et génétiques d’une maladie rare : la pseudo-hypoparathyroïdie (PHP) qui affecte l’homéostasie phosphocalcique par défaut de production d’AMPc (3’-5’-cyclique adénosine monophosphate) en réponse à la parathormone (PTH). Il s’agit d’une histoire aux multiples facettes, dont les bases moléculaires ont été progressivement dévoilées au fur et à mesure des progrès scientifiques, réunissant au sein d’un même locus, appelé GNAS (guanine nucleotide-binding protein alpha stimulating) qui code la sous-unité alpha stimulatrice de la protéine G (Gαs), marquée par un spectre d’affections que la clinique seule n’associerait pas. Différents mécanismes d’altération génétique, leur localisation sur le chromosome d’origine maternelle ou paternelle, vont en effet conduire à différentes maladies.
Un peu d’histoire
C’est sous le nom d’un curieux éponyme faisant référence à une vieille race de poules naines créées par Sir John Sebright, au xix e siècle, que l’endocrinologue américain Fuller Albright (1900-1969) décrivit pour la première fois en 1942 le « Seabright bantam syndrome », un syndrome morphologique caractérisé par une petite taille, une obésité, un faciès arrondi, des anomalies de longueur des doigts, accompagné souvent d’un retard mental, associé à une hypocalcémie et une hyperphosphatémie [1]. L’absence d’élévation de l’excrétion urinaire de phosphate, après administration d’extraits de glande parathyroïde [2], suggérait une résistance périphérique à la PTH plutôt qu’une insuffisance de production de l’hormone, d’où le nom de pseudohypoparathyroïdie. Cette description fut reprise par le pédiatre américain Charles Upton Lowe (1921-2012) en 1950, chez un patient qui présentait de plus une hypothyroïdie et des zones d’hypercalcification au niveau des poignets [3]. Le syndrome dysmorphique fut ultérieurement décrit sous le nom d’ostéodystrophie héréditaire d’Albright (OHA). Une décennie plus tard, Albright identifia un patient présentant les mêmes caractères dysmorphiques, mais sans anomalie phosphocalcique et une réponse appropriée à la PTH. Ce phénotype fut alors dénommé pseudo-pseudohypoparathyroïdie (PPHP) [4]. Le décryptage des mécanismes physiopathologiques de la maladie a ensuite suivi l’évolution des connaissances en biochimie puis en génétique.
L’action de la PTH sur le rein et sur l’os repose sur l’activation de l’adénylate cyclase, une enzyme qui transforme l’ATP en AMPc. Le dosage de la PTH, rendu possible par la technique radio-immunologique (radioimmunoassay, RIA), a permis de révéler chez les patients une élévation de la concentration plasmatique de la PTH, un maintien de la régulation de la concentration de la PTH par le calcium, et une absence d’excrétion urinaire d’AMPc en réponse à l’administration de PTH [5].
Les mécanismes de couplage entre un récepteur à sept domaines transmembranaires et la production d’AMPc via l’adénylate cyclase (AC) ont été disséqués au cours des années 1970, conduisant à l’identification d’un complexe protéique liant des nucléotides à guanine, qui a été appelé protéine G. Lorsque l’effecteur enzymatique est l’AC, ce complexe protéique porte le nom de protéine Gs. La PTH agit sur ses tissus cibles en se liant à PTHR1, un récepteur qui appartient à cette famille de récepteurs membranaires à sept domaines transmembranaires couplés aux protéines G (RCPG), récepteur qui lie la PTH et le PTH-rp (PTH related-peptide), un peptide homologue dans sa partie N-terminale avec la PTH.
Une fois éliminée l’hypothèse d’un dysfonctionnement de l’AC [6], il fut démontré que la maladie était liée à une baisse d’activité de la protéine Gs grâce à la mise au point d’un test fondé sur la complémentation entre les membranes d’érythrocytes humains (dépourvus d’AC) et celles d’érythrocytes de dinde (dépourvus de protéine Gs), ayant comme résultat une réduction d’environ 50 % de la production d’AMPc [7], une diminution retrouvée chez les patients atteints de PHP ou de PPHP. Ce test fut, par la suite, utilisé pour le diagnostic de la maladie [8].
La protéine Gs est un complexe hétérotrimérique qui est intermédiaire entre un RCPG membranaire et un effecteur enzymatique cytoplasmique, mais lié à la membrane, qui sera à l’origine de la synthèse d’un second messager intracellulaire. La protéine Gs est constituée de trois sous-unités : Gαs, Gβ et Gγ [9]. La sous-unité Gαs établit des contacts spécifiques avec l’AC et conduit à la production d’AMPc à partir de l’ATP. Elle fonctionne selon un mode on/off (Figure 1A, B).
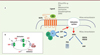 |
Figure 1. La sous-unité Gαs dans la signalisation intracellulaire. A. Lorsqu’un récepteur couplé aux protéines G à sept domaines transmembranaires (RCPG) fixe un ligand, tel qu’une hormone (PTH : parathormone ; PTH-rp : PTH-related peptide ; TSH : hormone thyréotrope ; GHRH : growth hormone-releasing hormone, etc.), il subit une modification de conformation lui permettant d’interagir avec la protéine G qui est constituée de trois sous-unités (Gα, Gβ et Gγ). Gα, liée à du GDP (Gα-GDP) dans sa forme inactive, libère alors le GDP et se lie à du GTP, tout en se dissociant des sous-unités Gβ et Gγ. Gα-GTP active ensuite l’adénylate cyclase (AC) pour produire de l’AMPc à partir d’ATP. La transduction du signal est assurée par une cascade de phosphorylations intracellulaires qui implique en particulier la protéine kinase A (PKA). La durée du signal est régulée par des phosphodiestérases, telles que PDE4D et PDE3A, qui métabolisent l’AMPc. R : unité régulatrice de la PKA (PRKAR1A) ; C : unité catalytique de la PKA. B. L’activation de l’adénylate cyclase est autolimitée dans le temps par l’activité GTPase intrinsèque de la sous-unité Gαs, éliminant le phosphate (Pi) en position g, traduisant un système de régulation de type « on / off ». Gαs, de nouveau liée à du GDP, reprend alors une forme inactive (Gα-GDP) et se réassocie avec les sous-unités Gβ et Gγ. |
Avec les progrès de la génétique moléculaire, le gène qui code la sous-unité Gαs a pu être identifié et caractérisé. Appelé GNAS1, il est localisé dans le locus GNAS situé dans la région télomérique du bras long du chromosome 20 (20q13.32). Ce gène est transcrit sous la forme d’un ARNm comprenant 13 exons codant une protéine de 394 acides aminés.
À partir de la détermination de la séquence du gène, les premières mutations de GNAS1 associées à la PHP ont pu être identifiées. Il s’agissait de mutations hétérozygotes, responsables d’une perte de fonction (LOF pour loss-of-function). Ces mutations peuvent être de différents types et sont répertoriées dans des banques de données (http://www.hgmd.cf.ac.uk/ac/index.php ou www.lovd.nl/GNAS). Elles peuvent être héritées ou survenir de novo. Chez la souris, les mutations LOF bi-alléliques sont létales. Dans un travail récent de notre équipe, il a été montré que les mutations conduisant à une perte de fonction de la sous-unité Gαs sont transmises préférentiellement à la descendance, suggérant ainsi un avantage sélectif dans la sélection des gamètes et/ou dans l’embryogenèse [10].
L’hétérozygotie pour la mutation permet donc d’expliquer l’haplo-insuffisance que l’on observe dans les érythrocytes par le test de complémentation (avec une réduction de 50 % de l’activité de Gs, voir plus haut). Cependant, les mécanismes conduisant à la résistance à la PTH au niveau rénal sont restés longtemps incompris.
Une altération des effecteurs en aval de Gαs, tels que PRKAR1A (protein kinase cAMP-dependent type I regulatory subunit alpha), et les phosphodiestérases PED3A et PED4D peuvent affecter la production de l’AMPc intracellulaire. Pourtant, les lésions identifiées sur les gènes codant ces effecteurs sont associées à des phénotypes différents de ceux observés dans la PHP entrant dans le registre des acrodysostoses, que nous ne développerons pas dans le cadre de cette revue [11].
Les différents types de PHP (Tableau I)
En parallèle des découvertes permettant de comprendre les mécanismes impliqués dans la maladie, le spectre pathologique de la PHP a été redéfini. Différents types ont ainsi été déterminés selon des caractéristiques qui leur sont spécifiques.
Les différents types de PHP. (+) Association ; OHA : ostéodystrophie héréditaire d’Albright ; PTH : parathormone ; TSH : hormone thyréotrope; OMIM : online mendelian inheritance in man (https://www.omim.org).
Ainsi, dans la PHP de type 1 (PHP1), l’administration de PTH exogène [2] ne produit ni élévation de l’AMPc ni phosphaturie. Ce type correspond à un défaut de la sous-unité Gαs. Dans le type 2 (PHP2), que nous n’aborderons pas ici, l’administration de PTH exogène élève le niveau d’AMPc urinaire, mais ne produit pas de phosphaturie, révélant un déficit situé en aval de la sous-unité Gαs [12].
L’ostéodystrophie héréditaire d’Albright (OHA) rassemble, quant à elle, plusieurs traits phénotypiques particuliers : une petite taille, une obésité, des anomalies des extrémités, telles que brachydactylie, brachymétacarpie et des ossifications sous-cutanées plus ou moins diffuses ; un retard mental est souvent présent.
La PHP de type 1A (PHP1A) regroupe des patients souffrant de PHP1 associée à une OHA avec une diminution de l’activité de la sous-unité Gαs.
La PHP de type 1B (PHP1B) regroupe des patients souffrant de PHP1 mais sans OHA et avec une activité Gαs quasi normale.
Enfin, la PPHP regroupe les patients atteints d’OHA avec baisse de l’activité de la sous-unité Gαs mais sans PHP. L’hétéroplasie progressive osseuse (POH) est une forme sévère de PPHP. Elle se caractérise par une ossification hétérotopique qui s’étend progressivement, en profondeur dans les tissus conjonctif et dans les muscles squelettiques [13].
Certains patients qui présentent des signes cliniques de PHP1 avec un syndrome dysmorphique mais sans baisse de l’activité de la sous-unité Gαs sont classés en PHP1C.
Les signes cliniques et biologiques de ces affectations ainsi que leur prise en charge ont fait l’objet de nombreuses publications et d’un consensus international [14].
La recherche d’anomalies génétiques, grâce au développement des séquenceurs nouvelle génération qui permettent l’étude en parallèle de panels de gènes, voire de l’exome, a pris le pas sur l’étude biochimique de l’activité Gs. Aussi, certains auteurs ont proposé une nouvelle classification : l’iPPSD, pour inactivating PTH/PTHrP signaling disorder, fondée sur le mécanisme commun de résistance à la PTH et sur la nature de la lésion génétique impliquée [15].
Des paradoxes cliniques éclairés par la biochimie et la génétique
Des études de cristallographie, de biochimie, ainsi que les signes cliniques et biologiques observés chez des patients porteurs de mutations touchant le gène GNAS1 ont permis de préciser des déterminants structuraux responsables d’effets spécifiques, perte de fonction ou gain de fonction [9, 16-18].
Des mutations perte de fonction : la PHP1C élucidée
Dans la PHP1C, une mutation correspondant à des modifications touchant l’un des cinq derniers acides aminés de la protéine n’affecte ni la transcription du gène ni la synthèse de la protéine Gαs. La protéine produite est cependant tronquée et ne peut se lier au récepteur RCPG. Malgré un tableau de PHP1A témoignant du défaut de Gαs, le test de complémentation in vitro, qui étudie le couplage avec l’AC, est donc, dans ce cas, normal [19].
Des mutations gain de fonction
La substitution, dans la protéine, de l’arginine en position 201 (Arg201) par une histidine ou une cytosine, ou moins fréquemment de la glycine en position 227 par une arginine, réduit l’échange du GTP par le GDP et donc le passage de l’état on vers l’état off de la protéine. Cela entraîne une activation constitutive des récepteurs et donc une production inappropriée d’AMPc, alors en excès. Retrouvée à l’état de mosaïque (ou mosaïcisme), cette mutation est à l’origine du syndrome de McCune-Albright [17] que nous ne developperons pas dans le cadre de cette revue.
De telles mutations sont retrouvées dans certaines tumeurs : tumeurs somatotropes, adénome thyroidien, etc. Elles sont alors appelées oncogène gsp. Notons enfin que la sous-unité Gαs comportant une arginine en position 201, est la cible de la toxine sécrétée par le vibrion cholérique, avec, pour effet délétère, une diarrhée sécrétoire.
Le locus GNAS est soumis à empreinte parentale
L’haploinsuffisance résultant de mutations retrouvées à l’état hétérozygote peut-elle, à elle seule, expliquer la résistance rénale à la PTH ?
Au sein d’une même famille, certains membres portant la même mutation peuvent présenter des phénotypes différents de PHP1A ou de PPHP. Les études de pédigrée intrafamilial montrent que les mutations de GNAS1, responsables de la PHP1A, sont héritées de la mère (ou situées sur l’allèle maternel si les mutations sont de novo). En revanche, ces mêmes mutations sont responsables de PPHP lorsqu’elles sont héritées du père (ou touchant l’allèle paternel) [20, 21]. L’expression de l’allèle maternel de GNAS1 est essentielle à la production d’AMPc urinaire résultant de l’action de la PTH, et une mutation sur l’allèle maternel entraîne une perte totale d’activité Gs et une résistance à la PTH.
D’une façon générale, l’empreinte génomique est un mécanisme selon lequel l’expression d’un gène est mono-allélique et non bi-allélique, l’autre allèle étant « éteint ». La modulation de la transcription est principalement due à une méthylation différentielle de certaines régions (DMR, pour differentially methylated regions) au niveau des promoteurs du gène : le gène dont le promoteur est méthylé n’est pas transcrit.
Comme la majorité des gènes soumis à empreinte génomique, le locus GNAS est organisé en cluster [21] (Figure 2A). L’épissage alternatif de l’exon 1 (GNAS contient 13 exons) donne naissance à trois transcrits qui codent différentes protéines : Gαs (exons 1-13) ; NESP55 (neuroendocrine secretory protein 55), qui fait partie de la famille des chromogranines et dont la séquence codante est entièrement contenue dans l’exon 1 ; XLas (isoforme extra-large) qui ne diffère de Gαs que par sa partie N-terminale et dont la distribution tissulaire est plus limitée (pancréas, cerveau, hypophyse, cœur, surrénales) [22]. Deux transcrits non codants sont également produits : A/B, et un ARN antisens AS (appelés respectivement 1A et nespas chez la souris).
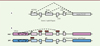 |
Figure 2. Organisation du locus GNAS. A. Épissage alternatif. Les exons 2 à 13 sont communs aux trois ARNm codant respectivement les protéines Gαs, XLas et NESP55 et à l’ARN non codant A/B. AS est un ARN non codant antisens. Chaque exon 1 spécifique est représenté par une boîte différente. Les flèches indiquent le sens de la transcription. B. Empreinte parentale. mat : allèle maternel ; pat : allèle paternel. ICR : centre de contrôle de la méthylation en cis de A/B DMR, situé dans le gène codant la syntaxine 16 (STX16). Les exons 1 spécifiques sont chacun sous le contrôle de promoteurs situés dans des DMR (région différentiellement méthylée). Les régions méthylées sur chacun des allèles sont représentées par des petits cercles rouge. Les DMR AS, XL et A/B sont méthylées sur l’allèle maternel alors que NESP55 DMR est méthylée sur l’allèle paternel. L’expression du gène codant Gαs est bi-allélique dans la plupart des tissus (érythrocytes, fibroblastes, etc.). Dans certains tissus (néphron, thyroïde, hypophyse, etc.), seul l’allèle maternel du gène codant Gαs est exprimé : l’empreinte est tissu-spécifique. |
Au locus GNAS, il existe quatre DMR : A/B DMR ; NESP55 DMR ; XL DMR et AS DMR. Les DMR spécifiques XL, AS et A/B, sont méthylées sur l’allèle maternel (Figure 2B) ; les transcrits dérivent donc de l’allèle paternel. Au contraire, NESP55 DMR est méthylée sur l’allèle paternel ; NESP55 s’exprime donc à partir de l’allèle maternel. Étonnament, il n’y a pas de DMR dans l’exon 1 spécifique de GNAS1 qui code Gαs, suggérant donc une expression bi-allélique. On constate cependant que lorsque A/B DMR est méthylée (A/B n’est donc pas exprimé), l’exon 1 spécifique de GNAS1 est utilisé, l’ARNm est transcrit puis traduit en Gαs. Lorsque A/B DMR n’est pas méthylée, c’est A/B qui est exprimé aux dépens de GNAS1. Chez la souris portant une mutation LOF de Gnas1 sur l’allèle maternel et présentant donc une PHP1A, la délétion de 1A sur l’allèle paternel permet l’expression de Gnas1 en cis, corrigeant ainsi le phénotype [23]. L’hypothèse du choix du promoteur libre par l’ARN polymérase est possible, mais elle ne peut expliquer pourquoi l’expression mono-allélique d’origine maternelle ne concerne que certains tissus et reste bi-allélique dans d’autres.
Une empreinte tissu-spécifique
Un défaut de signalisation touchant la protéine Gαs devrait concerner la majorité des RCPG utilisant la voie de l’AMPc. Or, la résistance à la PTH et, de façon moins constante, à la TSH (thyroid-stimulating hormone) responsable d’une hypothyroïdie, sont les principaux signes de la PHP1A associée à une perte de fonction hétérozygote sur l’allèle maternel. Les autres voies métaboliques impliquant la GH (growth hormone), la calcitonine, LH (luteinizing hormone)/FSH (follicle stimulating hormone), etc. sont en effet modérément affectées.
Dans les érythrocytes, l’expression de GNAS1 est bi-allélique : la baisse de 50 % de l’activité Gs chez les patients atteints de PHP1A ou de PPHP témoigne d’une haplo-insuffisance et non d’une perte de fonction complète. Chez la souris, des études d’immuno-histochimie ont montré qu’au niveau rénal, l’empreinte n’était pas identique dans les différentes parties du néphron : cette empreinte est présente dans le tube proximal mais pas dans la zone médullaire et le tube collecteur [24]. L’hypothèse de la synthèse locale d’un répresseur n’a cependant pas été confirmée, et les mécanismes conduisant à la mise en place d’une empreinte tissu-spécifique restent encore inconnus. Celle-ci s’installe progressivement après la naissance [25] : élévation de la PTH, hyperphosphatémie et enfin hypocalcémie.
La pseudo-hypoparathyroïdie et ses variantes
Gαs, remodelage osseux et qualité de l’os
La brachydactylie est l’anomalie la plus commune de l’ostéodystrophie héréditaire d’Albright (OHA). Cette atteinte touche préférentiellement le quatrième et le cinquième rayon du métacarpe et du métatarse. Elle résulte du défaut d’action du peptide PTH-rp sur les chondrocytes, entraînant l’accélération de la fermeture des plaques de croissance. Ce défaut concerne aussi les os longs, ce qui expliquerait, au moins en partie, la petite taille des patients [26].
Les ossifications (et non des calcifications) sous-cutanées font partie du phénotype d’OHA. De taille et en nombre variables, elles peuvent être particulièrement sévères et invalidantes : dans l’osteoma cutis, elles forment en effet de véritables plaques dermiques qui progressent en largeur selon une distribution en mosaïque, asymétrique avec une latéralisation qui suit le profil du dermomyotome1 ; dans l’hétéroplasie progressive osseuse (POH), l’ossification intramembraneuse suit un développement extensif et progressif vers les tissus profonds, les fascias 2 et les muscles. Ce caractère morbide est plus fréquemment observé chez des patients dont le gène GNAS1 comporte des mutations sévères (délétions, mutation tronquante, etc.) qui affectent plus particulièrement l’allèle paternel [13, 27]. La grande variabilité du processus d’ossification rend ainsi le conseil génétique particulièrement délicat. Certaines formes particulièrement invalidantes pourraient être liées à un mosaïcisme segmentaire de type 2, qui correspond à une mutation post-zygotique, secondaire à un allèle muté hérité d’un parent hétérozygote [28].
L’AMPc joue un rôle régulateur critique dans la détermination de l’orientation ostéoblastique ou adipocytaire des cellules. Il réduit en effet la voie d’activation des BMP (bone morphogenetic protein), ce qui inhibe la différenciation des ostéoblastes et donc favorise l’adipogenèse. L’AMPc aurait pour cible le morphogène Hedgehog (SHH) dont il diminuerait l’expression. L’haploinsuffisance de Gαs dans les progéniteurs mésenchymateux entraînerait une surexpression de la signalisation de SHH. Elle favoriserait ainsi la différenciation en ostéoblastes au détriment de l’adipogenèse [26, 29].
Obésité et retard de croissance in utero
Deux phénotypes opposés sont observés selon l’allèle parental affecté par une mutation perte de fonction. Présente dès la naissance, l’obésité fait partie du tableau clinique de la PHP1A. Son origine est multifactorielle, associant une résistance à l’action lipolytique de l’épinéphrine et un déficit de la voie activée par le récepteur de la mélanocortine (MC4R), qui contrôle la consommation énergétique et la prise alimentaire [18]. Une étude de re-séquencage3 a révélé des mutations de GNAS1 chez 2 % des enfants obèses. La caractérisation fonctionnelle, réalisée in vitro sur des cellules HEK (cellules embryonnaires de rein humain), a montré un degré de résistance variable en fonction des récepteurs étudiés. Un seuil critique de baisse d’activité Gs pourrait ainsi expliquer des tableaux cliniques variables et pauci-symptomatiques [18].
Les patients atteints de PPHP sont, au contraire, minces. Ils ont tous souffert d’un retard de croissance in utero, associé à une hypotrophie placentaire, d’autant plus important que la mutation affecte non seulement la sous-unité Gαs mais aussi XLas (exons 2 à 13 communs) [30]. Cette observation suggère que XLas, comme d’autres protéines codées par des gènes d’expression paternelle, jouerait un rôle dans la trophicité placentaire et la croissance fœtale.
PHP1B et épimutation
Les patients atteints de PHP1B présentent une hypocalcémie résistante à la PTH, avec peu ou pas de signes d’OHA et une activité érythrocytaire Gs normale. Une obésité morbide est souvent observée au cours des premières années de vie [14].
Il existe deux formes de PHP1B : une forme familiale de transmission autosomique dominante, AD-PHP1B, et une forme sporadique, Spor-PHP1B. Ces deux formes ont en commun une perte de méthylation (LOM) de la région différentiellement méthylée (DMR) de A/B (A/B DMR) [31] (Tableau II). La méthylation de A/B DMR dépend d’un centre d’empreinte qui est situé vers le centromère dans le gène de la syntaxine 16 (STX16) [32] à 220 kilobases (kb) du gène GNAS1.
Profils de méthylation des DMR observés dans les PHP1B. m : expression maternelle ; p : expression paternelle ; AD-PHP1B : forme autosomique dominante ; Spor-PHP1B : forme sporadique. L’expression de chaque gène est indiquée par (+) et la méthylation des DMR (région différentiellement méthylée) par (CH3). Dans la PHP1B, l’absence de méthylation de A/B DMR conduit à son expression par les deux allèles (+/+) et GNAS1 n’est pas exprimé.
De façon récurrente, dans l’AD-PHP1B, une délétion en cis de 3 kb est retrouvée dans le gène STX16 ; mais il existe d’autres mutations, comme des duplications ou des inversions. Chez la souris, les délétions dans le gène Stx16 n’affectent pas, par contre, la méthylation de 1A DMR (qui correspond à A/B DMR chez l’homme) ce qui limite l’intérêt de ce modèle. L’existence de petites délétions dans le gène NESP55, responsables d’AD-PHP1B, montre que la transcription de NESP55 semble également nécessaire à la méthylation de A/B DMR sur l’allèle maternel [33]. Cette régulation est confirmée chez la souris puisque la transcription ectopique sur l’allèle paternel de Nesp55 entraîne la méthylation de 1A DMR en cis [34]. Enfin, sur l’allèle paternel, la transcription de Nespas apparaît importante pour la méthylation de nesp DMR (ce qui éteint l’expression de Nesp) et pour prévenir la méthylation de 1A DMR (ce qui a pour conséquence l’expression de 1A et donc l’absence d’expression de Gnas1). Ce jeu de régulations complexes, organisant les DMR dans le locus GNAS, reste encore à approfondir. Les mécanismes mis en jeu feraient intervenir l’ouverture de la chromatine, ce qui faciliterait la mise en place de l’empreinte [35].
Les anomalies de méthylation observées dans les formes sporadiques sont plus complexes et leur mécanisme largement inconnu avec la perte de méthylation des DMR maternelles de AS, XL et A/B, et le gain de méthylation de NESP DMR. Une perte de l’allèle maternel ou une disomie4 ou hétérodisomie5 paternelle (patUPD20q), peuvent expliquer un profil typique de l’épigénome paternel [36]. Leur fréquence augmente avec l’âge de la mère [36]. Cependant, chez la majorité des patients, les anomalies de méthylation sont incomplètes, avec des gains et des pertes partiels de méthylation. Leur cause reste inconnue. Des défauts de méthylation sur d’autres locus soumis à empreinte (multi-locus imprinting defect) ont été observés [37], suggérant l’altération d’un réseau de gènes qui serait impliqué dans la mise en place de l’empreinte génomique [38].
Dans le spermatozoïde, l’empreinte est acquise avant la naissance. Dans l’ovocyte, les DMR de AS et XL se mettent en place après la naissance, pendant la phase de croissance. Après la fécondation, les DMR des gènes soumis à empreinte ne subissent pas la vague de dé-méthylation observée en période pré-implantatoire. L’asymétrie épigénétique persistera, préservant ainsi l’identité parentale [35]. Il est donc possible qu’une anomalie au sein de l’ovocyte soit responsable d’un défaut de la méthylation du locus GNAS responsable de la Spor-PHP1B. Bien que sa cause reste actuellement inconnue, elle pourrait être favorisée par les techniques de fécondation in vitro [39]. Des anomalies de méthylation peuvent également se trouver réparties en mosaïque, suggérant, dans ce cas, un événement post-zygotique plus tardif. Enfin, les jumeaux monozygotes atteints de spor-PHP1B, concordants sur le plan épigénétique, s’avèrent rares et les modifications épigénétiques que l’on observe chez ces patients impliquent une altération très précoce du processus [40].
Conclusion
Les pseudo-hypoparathyroïdies (PHP), dans leurs différentes formes, sont des maladies rares qui affectent la voie de la signalisation impliquant la sous-unité Gαs responsable de la production d’AMPc intracellulaire. Si la génétique moléculaire permet de nommer la maladie, de nombreuses interrogations persistent quant aux mécanismes conduisant aux différents phénotypes identifiés. L’altération de la séquence codante de GNAS1 avec perte de fonction de la protéine Gαs identifiée dans la PHP1A, ne donne pas le même phénotype qu’une épimutation qui altère l’expression génique de GNAS1, qui est retrouvée dans la PHP1B. Pour corriger au plus tôt les déficiences hormonales, un diagnostic précoce, le plus souvent génétique, se justifie. En revanche, la complexité des interactions entre les différentes formes cliniques de la maladie, lésions osseuses invalidantes, obésité, petite taille, troubles du développement, rend la définition de pistes thérapeutiques difficile pour traiter ces autres symptômes.
Liens d’intérêt
L’auteure déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Le dermomyotome est un feuillet épithélial transitoire qui se développe à partir du domaine dorsal du somite. Le dermomyotome produit une variété de types cellulaires qui participent à la formation des muscles, du derme dorsal et des vaisseaux sanguins.
Tissus fibreux qui enveloppent les muscles, les organes, les os, les nerfs et les vaisseaux.
Cette dénomination, essentiellement utilisée en génétique, désigne le séquençage d’un grand fragment d’ADN suivi de la comparaison du résultat obtenu avec celui d’une séquence de référence connue.
Anomalie caractérisée par le fait que deux chromosomes homologues proviennent d’un même parent.
Anomalie caractérisée par le fait que deux chromosomes homologues, non identiques, proviennent d’un même parent.
Références
- Albright F, Burnett CH, Smith P, Parson W. Pseudohypoparathyroidism, An example of “Seabright Bantam Syndrome”. Report of Three Cases. Endocrinology 1942 ; 30 : 922–932. [Google Scholar]
- Ellsworth R, Howard JE. Studies on the physiology of the parathyroid glands. Some re- sponses of normal human kidneys and blood to intravenous parathyroid extract. Bull Johns Hopkins Hosp 1934 ; 55 : 296. [Google Scholar]
- Lowe CU, Ellinger AJ, Wright W, Stauffer H. Pseudohypoparathyroidism; the Seabright bantam syndrome. J Pediatr 1950 ; 36 : 1–10. [CrossRef] [PubMed] [Google Scholar]
- Albright F, Forbes AP, Henneman PH. Pseudo-pseudohypoparathyroidism. Trans Assoc Am Physicians 1952 ; 65 : 337–350. [PubMed] [Google Scholar]
- Chase LR, Melson GL, Aurbach GD. Pseudohypoparathyroidism: defective excretion of 3’,5’-AMP in response to parathyroid hormone. J Clin Invest 1969 ; 48 : 1832–1844. [CrossRef] [PubMed] [Google Scholar]
- Farfel Z, Brickman AS, Kaslow HRet al. Defect of receptor-cyclase coupling protein in pseudohypoparathyroidism. N Engl J Med 1980 ; 303 : 237–242. [CrossRef] [PubMed] [Google Scholar]
- Levine MA, Downs RW, Singer Met al. Deficient activity of guanine nucleotide regulatory protein in erythrocytes from patients with pseudohypoparathyroidism. Biochem Biophys Res Commun 1980 ; 94 : 1319–1324. [CrossRef] [PubMed] [Google Scholar]
- Marguet C, Mallet E, Basuyau JPet al. Clinical and biological heterogeneity in pseudohypoparathyroidism syndrome. Results of a multicenter study. Horm Res 1997 ; 48 : 120–130. [CrossRef] [PubMed] [Google Scholar]
- Hamm HE. The many faces of G protein signaling. J Biol Chem 1998 ; 273 : 669–672. [CrossRef] [PubMed] [Google Scholar]
- Snanoudj S, Molin A, Colson, et al. Maternal Transmission Ratio Distortion of GNAS Loss-of-Function Mutations. J Bone Miner Res 2020; 35 : 913–9. [CrossRef] [PubMed] [Google Scholar]
- Linglart A, Menguy C, Couvineau Aet al. Recurrent PRKAR1A mutation in acrodysostosis with hormone resistance. N Engl J Med 2011 ; 364 : 2218–2226. [CrossRef] [PubMed] [Google Scholar]
- Drezner M, Neelon FA, Lebovitz HE. Pseudohypoparathyroidism type II: a possible defect in the reception of the cyclic AMP signal. N Engl J Med 1973 ; 289 : 1056–1060. [CrossRef] [PubMed] [Google Scholar]
- Shore EM, Ahn J, Jan de Beur Set al. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N Engl J Med 2002 ; 346 : 99–106. [CrossRef] [PubMed] [Google Scholar]
- Mantovani G, Bastepe M, Monk Det al. Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement. Nat Rev Endocrinol 2018 ; 14 : 476–500. [CrossRef] [PubMed] [Google Scholar]
- Thiele S, Mantovani G, Barlier Aet al. From pseudohypoparathyroidism to inactivating PTH/PTHrP signalling disorder (iPPSD), a novel classification proposed by the EuroPHP network. Eur J Endocrinol 2016 ; 175 : 1–17. [Google Scholar]
- Lambright DG, Noel JP, Hamm HE, Sigler PB. Structural determinants for activation of the alpha-subunit of a heterotrimeric G protein. Nature 1994 ; 369 : 621–628. [CrossRef] [PubMed] [Google Scholar]
- Weinstein LS, Liu J, Sakamoto Aet al. Minireview: GNAS: normal and abnormal functions. Endocrinology 2004 ; 145 : 5459–5464. [CrossRef] [PubMed] [Google Scholar]
- Mendes de Oliveira E, Keogh JM, et al. Obesity-Associated GNAS Mutations and the Melanocortin Pathway. N Engl J Med 2021; 385 : 1581–92. [CrossRef] [PubMed] [Google Scholar]
- Thiele S, de Sanctis L, Werner Ret al. Functional characterization of GNAS mutations found in patients with pseudohypoparathyroidism type Ic defines a new subgroup of pseudohypoparathyroidism affecting selectively Gs-alpha-receptor interaction. Hum Mutat 2011 ; 32 : 653–660. [CrossRef] [PubMed] [Google Scholar]
- Davies SJ, Hughes HE. Imprinting in Albright’s hereditary osteodystrophy. J Med Genet 1993 ; 30 : 101–103. [CrossRef] [PubMed] [Google Scholar]
- Hayward BE, Moran V, Strain L, Bonthron DT. Bidirectional imprinting of a single gene: GNAS1 encodes maternally, paternally, and biallelically derived proteins. Proc Natl Acad Sci U S A 1998 ; 95 : 15475–15480. [CrossRef] [PubMed] [Google Scholar]
- Bastepe M, Gunes Y, Perez-Villamil Bet al. Receptor-mediated adenylyl cyclase activation through XLalphas, the extra-large variant of the stimulatory G Protein alpha-Subunit. Mol Endocrinol 2002 ; 16 : 1912–1919. [CrossRef] [PubMed] [Google Scholar]
- Williamson CM, Ball ST, Nottingham WTet al. A cis-acting control region is required exclusively for the tissue-specific imprinting of Gnas. Nat Genet 2004 ; 36 : 894–899. [CrossRef] [PubMed] [Google Scholar]
- Weinstein LS, Yu S, Ecelbarger CA. Variable imprinting of the heterotrimeric G protein G(s) alpha-subunit within different segments of the nephron. Am J Physiol Renal Physiol 2000 ; 278 : F507–F514. [CrossRef] [PubMed] [Google Scholar]
- Turan S, Fernandez-Rebollo E, Aydin Cet al. Postnatal establishment of allelic Gαs silencing as a plausible explanation for delayed onset of parathyroid hormone resistance owing to heterozygous Gαs disruption. J Bone Miner Res 2014 ; 29 : 749–760. [CrossRef] [PubMed] [Google Scholar]
- Cong Q, Xu R, Yang Y. Gαssignaling in skeletal development, homeostasis and diseases Curr Top Dev Bio 2019; 133 : 281–307. [CrossRef] [Google Scholar]
- Lebrun M, Richard N, Abeguilé Get al. Progressive osseous heteroplasia: a model for the imprinting effects of GNAS inactivating mutations in humans. J Clin Endocrinol Metab 2010 ; 95 : 3028–3038. [CrossRef] [PubMed] [Google Scholar]
- Happle R.. Progressive osseous heteroplasia is not a Mendelian trait but a type 2 segmental manifestation of GNAS inactivation disorders: A hypothesis. Eur J Med Genet 2016 ; 59 : 290–294. [CrossRef] [PubMed] [Google Scholar]
- Regard JB, Malhotra D, Gvozdenovic-Jeremic Jet al. Activation of Hedgehog signaling by loss of GNAS causes heterotopic ossification. Nat Med 2013 ; 19 : 1505–1512. [CrossRef] [PubMed] [Google Scholar]
- Richard N, Molin A, Coudray Net al. Paternal GNAS mutations lead to severe intrauterine growth retardation (IUGR) and provide evidence for a role of XLαs in fetal development. J Clin Endocrinol Metab 2013 ; 98 : 1549–1556. [Google Scholar]
- Liu J, Litman D, Rosenberg MJet al. A GNAS1 imprinting defect in pseudohypoparathyroidism type IB. J Clin Invest 2000 ; 106 : 1167–1174. [CrossRef] [PubMed] [Google Scholar]
- Bastepe M, Fröhlich LF, Hendy GNet al. Autosomal dominant pseudohypoparathyroidism type Ib is associated with a heterozygous microdeletion that likely disrupts a putative imprinting control element of GNAS. J Clin Invest 2003 ; 112 : 1255–1263. [CrossRef] [PubMed] [Google Scholar]
- Rezwan FI, Poole RL, Prescott Tet al. Very small deletions within the NESP55 gene in pseudohypoparathyroidism type 1b. Eur J Hum Genet 2015 ; 23 : 494–499. [CrossRef] [PubMed] [Google Scholar]
- Mehta S, Williamson CM, Ball Set al. Transcription driven somatic DNA methylation within the imprinted Gnas cluster. PLoS One 2015 ; 10 : e0117378. [CrossRef] [PubMed] [Google Scholar]
- Zeng Y, Chen T. DNA Methylation reprogramming during mammalian development. Genes 2019 ; 10 : 257. [CrossRef] [Google Scholar]
- Colson C, Decamp M, Gruchy Net al. High frequency of paternal iso or heterodisomy at chromosome 20 associated with sporadic pseudohypoparathyroidism 1B. Bone 2019 ; 123 : 145–152. [CrossRef] [PubMed] [Google Scholar]
- Maupetit-Méhouas S, Azzi S, Steunou Vet al. Simultaneous hyper- and hypomethylation at imprinted loci in a subset of patients with GNAS epimutations underlies a complex and different mechanism of multilocus methylation defect in pseudohypoparathyroidism type 1b. Hum Mutat 2013 ; 34 : 1172–1180. [CrossRef] [PubMed] [Google Scholar]
- Monteagudo-Sánchez A, Hernandez Mora JR, Simon C, et al. The role of ZFP57 and additional KRAB-zinc finger proteins in the maintenance of human imprinted methylation and multi-locus imprinting disturbances. Nucleic Acids Res 2020; 48 : 11394–407. [CrossRef] [PubMed] [Google Scholar]
- Milioto A, Reyes M, Hanna P, et al. Lack of GNAS re-methylation during oogenesis may be a cause of sporadic pseudohypoparathyroidism type Ib (PHP1B). J Clin Endocrinol Metab 2022; 107 : e1610–9. [CrossRef] [PubMed] [Google Scholar]
- Keidai Y, Iwasaki Y, Iwasaki K, et al. Sporadic Pseudohypoparathyroidism Type 1B in Monozygotic Twins: Insights Into the Pathogenesis of Methylation Defects. J Clin Endocrinol Metab 2022; 107 : e947–54. [CrossRef] [PubMed] [Google Scholar]
Liste des tableaux
Les différents types de PHP. (+) Association ; OHA : ostéodystrophie héréditaire d’Albright ; PTH : parathormone ; TSH : hormone thyréotrope; OMIM : online mendelian inheritance in man (https://www.omim.org).
Profils de méthylation des DMR observés dans les PHP1B. m : expression maternelle ; p : expression paternelle ; AD-PHP1B : forme autosomique dominante ; Spor-PHP1B : forme sporadique. L’expression de chaque gène est indiquée par (+) et la méthylation des DMR (région différentiellement méthylée) par (CH3). Dans la PHP1B, l’absence de méthylation de A/B DMR conduit à son expression par les deux allèles (+/+) et GNAS1 n’est pas exprimé.
Liste des figures
|
Figure 1. La sous-unité Gαs dans la signalisation intracellulaire. A. Lorsqu’un récepteur couplé aux protéines G à sept domaines transmembranaires (RCPG) fixe un ligand, tel qu’une hormone (PTH : parathormone ; PTH-rp : PTH-related peptide ; TSH : hormone thyréotrope ; GHRH : growth hormone-releasing hormone, etc.), il subit une modification de conformation lui permettant d’interagir avec la protéine G qui est constituée de trois sous-unités (Gα, Gβ et Gγ). Gα, liée à du GDP (Gα-GDP) dans sa forme inactive, libère alors le GDP et se lie à du GTP, tout en se dissociant des sous-unités Gβ et Gγ. Gα-GTP active ensuite l’adénylate cyclase (AC) pour produire de l’AMPc à partir d’ATP. La transduction du signal est assurée par une cascade de phosphorylations intracellulaires qui implique en particulier la protéine kinase A (PKA). La durée du signal est régulée par des phosphodiestérases, telles que PDE4D et PDE3A, qui métabolisent l’AMPc. R : unité régulatrice de la PKA (PRKAR1A) ; C : unité catalytique de la PKA. B. L’activation de l’adénylate cyclase est autolimitée dans le temps par l’activité GTPase intrinsèque de la sous-unité Gαs, éliminant le phosphate (Pi) en position g, traduisant un système de régulation de type « on / off ». Gαs, de nouveau liée à du GDP, reprend alors une forme inactive (Gα-GDP) et se réassocie avec les sous-unités Gβ et Gγ. |
| Dans le texte | |
|
Figure 2. Organisation du locus GNAS. A. Épissage alternatif. Les exons 2 à 13 sont communs aux trois ARNm codant respectivement les protéines Gαs, XLas et NESP55 et à l’ARN non codant A/B. AS est un ARN non codant antisens. Chaque exon 1 spécifique est représenté par une boîte différente. Les flèches indiquent le sens de la transcription. B. Empreinte parentale. mat : allèle maternel ; pat : allèle paternel. ICR : centre de contrôle de la méthylation en cis de A/B DMR, situé dans le gène codant la syntaxine 16 (STX16). Les exons 1 spécifiques sont chacun sous le contrôle de promoteurs situés dans des DMR (région différentiellement méthylée). Les régions méthylées sur chacun des allèles sont représentées par des petits cercles rouge. Les DMR AS, XL et A/B sont méthylées sur l’allèle maternel alors que NESP55 DMR est méthylée sur l’allèle paternel. L’expression du gène codant Gαs est bi-allélique dans la plupart des tissus (érythrocytes, fibroblastes, etc.). Dans certains tissus (néphron, thyroïde, hypophyse, etc.), seul l’allèle maternel du gène codant Gαs est exprimé : l’empreinte est tissu-spécifique. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.