")
")
| Issue |
Med Sci (Paris)
Volume 32, Number 6-7, Juin–Juillet 2016
|
|
|---|---|---|
| Page(s) | 625 - 633 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20163206027 | |
| Published online | 12 juillet 2016 | |
Itinéraire d’un agent double
NO, S-nitrosylation et cancer
NO and cancer: itinerary of a double agent
1
Université de Bourgogne Franche-Comté, LIIC EA7269, 7, boulevard Jeanne d’Arc, F-21000
Dijon, France
2
EPHE, PSL Research University, F-75014
Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La S-nitrosylation est une modification post-traductionnelle par laquelle le monoxyde d’azote (NO) régule l’activité des protéines. Cette modification particulière exerce un rôle déterminant dans le contrôle des mécanismes de transduction du signal qui gouvernent de nombreux processus cellulaires. Aujourd’hui, il apparaît que la S-nitrosylation joue un double jeu, activateur ou inhibiteur, dans la signalisation cellulaire de la croissance tumorale ou l’induction de la mort cellulaire. Grâce aux travaux démontrant l’effet cytotoxique du NO, de nouvelles stratégies thérapeutiques visant à utiliser des donneurs de NO ont été développées. Leur mode d’action, encore mal connu, cible un nombre important de protéines. Cette revue fait le point sur l’état actuel de nos connaissances sur les protéines S-nitrosylées de la signalisation oncogénique et apoptotique, en mettant en avant les protéines pour lesquelles la S-nitrosylation représente un intérêt thérapeutique pour le traitement du cancer.
Abstract
Protein S-nitrosylation is now recognized as a ubiquitous regulatory mechanism. Like any post-translational modifications, S-nitrosylation is critical for the control of numerous cellular processes. It is now clear that S-nitrosylation is playing a double game, enhancing or inhibiting the tumor growth or the induction of cell death. Thanks to research aimed at demonstrating NO cytotoxic effects, new therapeutic strategies based on NO donor drugs have emerged. Although therapeutic NO donors can target a large number of proteins, the cellular mechanism is still not fully understood. This review reflects the current state of knowledge on S-nitrosylated proteins that take part of the oncogenic and apoptotic signaling, putting forward proteins with potential interest in cancer therapy.
© 2016 médecine/sciences – Inserm
Vignette (Photo © Inserm-Sandrine Humbert et Morgane Thion).
Le rôle du monoxyde d’azote, ou NO (nitric oxide), dans la biologie du cancer est une histoire qui continue de soulever de nombreuses controverses. Le NO est une petite molécule gazeuse radicalaire, très réactive, connue pour jouer un rôle ambivalent, aussi bien antitumorigène que protumorigène. Au sein de la cellule, le NO est produit par une famille d’enzymes appelées NO-synthases (NOS) qui catalyse l’oxydation de la L-arginine en L-citrulline et en NO. Quatre NOS, la NOS neuronale (nNOS), la NOS inductible (iNOS), la NOS endothéliale (eNOS) et la NOS mitochondriale (mtNOS, l’isoforme α de la eNOS) assurent la production intracellulaire de NO aussi bien dans des conditions physiologiques que pathologiques. L’action biochimique du NO concerne essentiellement les protéines en provoquant des modifications post-traductionnelles (MPT) telles que la S-nitrosylation, la tyrosine nitration ou encore la métal S-nitrosylation. D’abord considérée comme une MPT atypique, la S-nitrosylation est à présent mieux décrite et reconnue pour être indispensable pour la régulation de la fonction de nombreuses protéines et voies de signalisation qui sont impliquées dans toutes les fonctions cellulaires. La S-nitrosylation constitue la MPT majeure, engendrée par le NO sur les protéines. Elle module leurs structures, leurs fonctions, leurs expressions, leurs localisations ou encore leurs interactions avec d’autres partenaires protéiques. La nature dichotomique du NO dans le cancer est dictée par l’impact de la S-nitrosylation sur des protéines impliquées aussi bien dans la survie que dans la mort cellulaire. Au cours de ces dernières années, plusieurs travaux s’accordent sur le fait que l’impact fonctionnel de la S-nitrosylation sur les protéines est largement dépendant du contexte cellulaire et de son statut rédox1.
La S-nitrosylation : une modification post-traductionnelle à part entière
Le NO est l’une des plus petites molécules endogènes impliquées dans des fonctions biologiques. Avec une demi-vie très courte, de l’ordre de la seconde, le monoxyde d’azote a une action restreinte aux protéines qui sont situées dans son environnement proche. La S-nitrosylation est la fixation non-enzymatique d’une molécule de NO sur un groupement thiol (-SH), porté par les cystéines. Le dérivé nitrosé (-SNO) ainsi formé présente toutefois une stabilité limitée dans le temps, puisqu’il peut subir des dénytrosylations qui font intervenir aussi bien des mécanismes non enzymatiques (processus de transnitrosylation) qu’enzymatiques (thioredoxines). Bien qu’obéissant à un processus moléculaire non-enzymatique, la S-nitrosylation des protéines est loin d’être un événement laissé au hasard. L’ensemble des groupements thiol libres d’une protéine ne font pas obligatoirement l’objet d’une S-nitrosylation. Si l’analyse séquentielle de nombreuses protéines S-nitrosylées a permis de déterminer des séquences consensus où les groupements thiol des cystéines les plus susceptibles de réagir avec un groupement NO sont contenus dans un environnement en acide aminés acides ou basiques, la structure tridimensionnelle de la protéine doit être néanmoins considérée. Toutefois, la spécificité du NO pour sa cible dépend, en réalité, de nombreux autres facteurs intrinsèques tels que la présence d’espèces réactives de l’oxygène (ERO ou ROS, en anglais) qui modulent la concentration intracellulaire de NO (l’ion superoxyde en réagissant avec le NO induit en effet la formation de peroxynitrites et une diminution du taux de NO), la localisation subcellulaire ou encore la durée d’exposition à la protéine cible. Le caractère dynamique, réversible, de ce processus revêt donc une importance primordiale dans la signalisation cellulaire.
Les MPT jouent un rôle capital dans les voies de signalisation cellulaire puisqu’elles sont au cœur des processus de régulation dynamique des fonctions biologiques des protéines. Pour bien mesurer l’impact de la S-nitrosylation sur les propriétés d’une protéine (et à plus grande échelle son impact sur une voie de signalisation), il est nécessaire de considérer l’importance que revêt un résidu cystéine au sein de sa structure (Figure 1). En effet, les résidus cystéine exercent un rôle charnière dans la structure, la stabilité, l’activité, la fonction et la localisation subcellulaire des protéines.
 |
Figure 1. La cystéine au cœur de la S-nitrosylation. S-nitrosylation : régulation de l’activité des protéines. De nombreuses enzymes impliquées dans la MPT (modification post-traductionelle) des protéines possèdent un résidu cystéine dans leur site catalytique. La réactivité du monoxyde d’azote (NO) avec un tel résidu abroge la plupart du temps leur activité. Les enzymes de types kinases (par exemple IKKβ [inhibitor of NF-kB kinase beta]), phosphatases (par exemple PTEN [phosphatase and tensin homolog]), E3 ubiquitine ligases (par exemple XIAP [X-linked inhibitor of apoptosis]) sont souvent la cible du NO au sein de différentes voies de signalisation contrôlant la survie ou la mort cellulaire. De ce fait, la S-nitrosylation d’une cystéine spécifique peut influencer la survenue d’un grand nombre de MPT et par conséquent altérer plusieurs voies de signalisation cellulaire aussi bien positivement que négativement. S-nitrosylation : régulation de la structure et stabilité des protéines. Les groupements thiols formant ensemble des ponts disulfures ne réagissent pas avec le NO et par conséquent ne constituent pas des sites de S-nitrosylation. Néanmoins deux sites S-nitrosylés peuvent a posteriori réagir entre eux pour former un pont disulfure (par exemple Akt1 [RAC-alpha serine/threonine-protein kinase]) contribuant ainsi à la stabilité de la structure ternaire et quaternaire de la protéine. En revanche, la S-nitrosylation peut perturber la stabilité d’une métalloprotéine qui met en jeu des résidus cystéines ayant des propriétés de liaison avec les métaux. La S-nitrosylation du facteur HIF1α (hypoxia inducible factor 1 alpha) entraîne sa stabilisation. S-nitrosylation : régulation de la fonction et localisation des protéines. La S-nitrosylation peut moduler le rôle d’une protéine lorsque les résidus cystéine ne sont impliqués ni dans le domaine catalytique d’une enzyme ni dans sa stabilité. La S-nitrosylation peut influencer la capacité d’une protéine à interagir avec un partenaire déterminé et contrecarrer sa fonction. Dans le cas de TSC (tuberous sclerosis complex)2, sa S-nitrosylation prévient son interaction avec son partenaire protéique TSC1 et ainsi sa fonction. Au contraire, la S-nitrosylation de cFLIP (cellular FLICE [FADD-like IL-1β-converting enzyme]-inhibitory protein) induit son interaction avec RIPK1 (receptor-interacting protein kinase 1) pour promouvoir sa capacité à induire la voie classique NF-kB (nuclear factor-kappa B) et/ou sa localisation subcellulaire en affectant par exemple les processus de transcription (la protéine GAPDH [glyceraldehyde-3-phosphate dehydrogenase] sous sa forme S-nitrosylée assure son interaction avec la protéine Siah1 [siah E3 ubiquitin protein ligase 1] qui induit sa translocation dans le noyau), de traduction, de dégradation des protéines ou encore de modifications post-traductionnelles. S-nitrosylation : sources de nouvelles MPT. Les propriétés du groupement thiol d’une cystéine en font un site privilégié pour de nombreuses autres MPT telles que la S-palmitoylation (par exemple Ras), la S-glutathionylation (par exemple caspase-3) ou encore la S-sulfhydration (par exemple GAPDH) [49]. Il est important de noter que la S-nitrosylation d’un site peut à la fois prévenir la formation d’une autre MPT ou au contraire servir d’intermédiaire pour la formation d’une nouvelle MPT oxidative selon l’environnement rédox dans lequel il se trouve. |
S-nitrosylation et régulation des voies de signalisation de prolifération cellulaire
L’activation inappropriée de nombreuses voies de signalisation impliquées dans la croissance, la survie cellulaire ou encore l’invasion est une caractéristique bien déterminée du processus tumoral. Parmi ces voies, les signalisations impliquant les MAPK (mitogen-activated protein kinases), PI3K/AKT (phosphatidylinositol-3-kinase/protein kinase B) et Wnt/β-caténine sont considérées comme des cibles majeures pour les thérapies anticancéreuses. À noter que plusieurs composants de ces voies de signalisation sont régulés par le NO (Figure 2).
 |
Figure 2. S-nitrosylation et principales voies de signalisation impliquées dans la prolifération cellulaire. La S-nitrosylation module l’activité (inhibition ou activation) de nombreux acteurs des voies de signalisation MAPK (mitogen-activated protein kinase) et PI3K/AKT (phosphatidylinositol-3-kinase/protein kinase B) initiées via les récepteurs à tyrosine kinase. La voie mTOR (mammalian target of rapamycin) joue un rôle clé dans l’équilibre entre survie et mort cellulaire. Il existe deux complexes protéiques fonctionnellement distincts formés par mTOR : mTORC1 (mammalian target of rapamycin complex 1) (associé entre autres à raptor) et mTORC2 (mammalian target of rapamycin complex 2) (associé entre autres à rictor). L’activation de la voie Wnt/β-caténine (wingless and int-1/beta-catenin) déstabilise le complexe de destruction de la β-caténine, APC (adenomatous polyposis coli), Axine, CKIα (casein kinase I a), GSK3β (glycogen synthase kinase 3). La S-nitrosylation de la β-caténine prévient non seulement son interaction avec le facteur de transcription TCF-4 (T cell transcription factor 4) mais sert aussi de signal précédant sa dégradation par le protéasome. L’ensemble des flèches correspond à la transduction du signal lorsque les voies sont normalement actives. MEK : mitogen-activated protein kinase kinase (ou MAP2K) ; ERK : extracellular signal-regulated kinases ; PKC : protéine kinase C ; SGK : serum-and glucocorticoid-induced protein kinase ; TSC : tuberous sclerosis complex ; Rheb : Ras homology enriched in brain ; PTEN : phosphatase and tensin homolog ; PIP2 : phosphatidylinositol-4,5-bisphosphate ; PIP3 : phosphatidylinositol (3,4,5)-trisphosphate ; Dsh : dishevelled ; LRP5/6 : low-density-lipoprotein receptor-related protein 5/6 ; S6K1 : S6 kinase 1 ; 4E-BP1 : 4E-binding protein 1 ; Ets-1 : E26 transformation-specific sequence-1. |
Voie classique MAPK ou voie ERK
Typiquement, la fixation du facteur de croissance EGF (epithelial growth factor) à son récepteur EGFR (epithelial growth factor receptor) permet l’initiation d’un signal, transmis depuis la membrane plasmique vers le noyau, qui facilite la prolifération cellulaire. Ce signal, initié par l’activité de petites protéines G (Ras ou p21Ras) et de nombreuses kinases associées au récepteur, est régulé par le NO à différents niveaux. Un effet antiprolifératif, observé à une concentration physiologique de NO dans des cellules de neuroblastome, induit la S-nitrosylation du récepteur EGFR sur les cystéines (principalement) en position 166 (Cys166) et 305 (Cys305) présentes dans son domaine extracellulaire. Celle-ci aurait vraisemblablement un effet inhibiteur en interférant avec la fixation du ligand sur son récepteur [1, 2]. Au contraire, dans le cancer du sein, négatif pour les récepteurs des estrogènes (RE-), la S-nitrosylation de l’EGFR, et de Src, les rend autonomes vis-à-vis des facteurs de croissance, en favorisant la stimulation de leur activité tyrosine kinase et la propagation d’une signalisation oncogénique [3]. Bien que l’ensemble de ces résultats apparaissent contradictoires, les sites exacts de la S-nitrosylation qui sont associés à l’activation de l’EGFR restent encore à déterminer. Il est en effet envisageable que la S-nitrosylation et l’activation de l’EGFR fasse intervenir des résidus cystéines différents de ceux préalablement identifiés en position 166 et 305, notamment dans la partie intracytoplasmique du récepteur, par exemple.
De la même façon, il a été montré dans un modèle utilisant des cellules de cancer du sein RE- que la S-nitrosylation de Ras sur la cystéine située en position 118 (Cys118) stimule l’échange de nucléotide guanine et conduit à la phosphorylation et à l’activation du facteur de transcription Est-1 par la cascade de signalisation Ras/MEK/ERK (Ras/mitogen-activated protein kinase/ERK kinase/extracellular-signal-regulated kinase) [4]. En réalité, il existe peu d’exemples pour lesquels la S-nitrosylation a un effet promoteur sur l’activité d’une enzyme. Plus fréquemment, la S-nitrosylation exerce un effet inhibiteur sur l’activité d’une enzyme en ciblant les résidus cystéine de leurs sites catalytiques. Certaines kinases, activées en aval par Ras, sont rendues inactives par S-nitrosylation ce qui peut constituer un frein à la propagation du signal oncogénique. Les protéines ERK1/2 sont une cible du NO. La S-nitrosylation de la cystéine 183 (Cys183) prévient la phosphorylation et conduit à la mort des cellules par apoptose [5].
Voie PI3K/AKT/mTOR
PI3K/AKT/mTOR (phosphatidylinositol-3-kinase/protein kinase B/mammalian target of rapamycin) est la voie de signalisation la plus fréquemment dérégulée dans les cancers solides humains [6]. Il s’agit d’une plate-forme de signalisation jouant un rôle prépondérant dans la régulation d’un grand nombre de fonctions cellulaires. Les nombreuses mutations de ses composants à l’origine de leurs délétions ou surexpressions favorisent le développement de tumeurs, l’angiogenèse et la chimiorésistance. L’étude intensive des mécanismes moléculaires régulant cette voie de signalisation a été associée à l’identification de nombreux inhibiteurs qui demeure un axe de recherche important dans le développement de thérapeutiques antitumorales. Ainsi, certains inhibiteurs de la protéine AKT continuent à faire l’objet de nombreux essais cliniques. La voie de signalisation PI3K/AKT/mTOR est activée de différentes façons : par la fixation de facteurs de croissance ou d’interleukines à leurs récepteurs, ou par l’interaction de PI3K avec p21Ras sous sa forme activée. Elle initie l’activation d’une cascade d’effecteurs en aval. Dans une lignée cellulaire de mélanome humain, le NO physiologique, produit par la iNOS (inducible nitric oxide synthase), favorise l’activation de la voie mTORC1 (mTOR complexe 1) via l’inactivation de son répresseur direct TSC1/TSC2 (tuberous sclerosis complex 1/2). La S-nitrosylation de TSC2 empêche sa dimérisation avec sa protéine partenaire TSC1 (en absence de cette hétérodimérisation, il y a une perte de la fonction de suppresseur de tumeur du complexe TSC1/TSC2), prévenant ainsi l’inhibition de mTORC1 [7]. PTEN (phosphatase and tensin homolog) est un important régulateur négatif de la voie PI3K/AKT/mTOR. Sous sa forme S-nitrosylée, au niveau de la cystéine en position 83 (Cys83), PTEN n’assure plus sa fonction de régulation et participe, en particulier, à l’angiogenèse tumorale [8]. Indépendamment du cancer, plusieurs autres travaux ont contribué à montrer l’effet inhibiteur du NO sur la voie PI3K/AKT/mTOR, notamment par la S-nitrosylation et l’inactivation de la protéine AKT [9, 10].
Voie Wnt/β-caténine/TCF
L’activation aberrante de la voie de signalisation Wnt/β-caténine est un évènement central dans les premières étapes de la transformation maligne des cellules coliques mais aussi dans le développement de nombreux cancers. L’effet antiprolifératif de l’aspirine NO (une forme d’aspirine associée au NO qui prévient la toxicité digestive de l’aspirine) qui est observé sur plusieurs lignées cellulaires coliques, mais aussi mammaires, est lié à l’inactivation de la voie Wnt/β-caténine. Le mécanisme d’action du NO conduisant à cet effet met en jeu la S-nitrosylation de la β-caténine qui prévient l’association et l’activation du facteur de transcription TCF-4 (T cell factor-4) et diminue l’expression de la cycline D1 dont la fonction (sous forme d’un complexe avec une kinase dépendante des cyclines) participe à la progression des cellules en début de phase G1 du cycle cellulaire [11, 12]. L’efficacité antitumorale de l’aspirine NO observée dans un modèle murin de leucémie lymphoblastique chronique est associée à une inactivation de la voie Wnt/ β-caténine par clivage et dégradation de la β-caténine [13].
S-nitrosylation et implication dans la mort cellulaire
Le processus d’homéostasie cellulaire est soumis en permanence à une régulation dynamique stricte placée sous le contrôle de voies de signalisation de survie et de mort cellulaires qui limite la prolifération de cellules potentiellement néfastes pour l’organisme. La S-nitrosylation des protéines contribue aussi bien à assurer le maintien que le déséquilibre de l’homéostasie cellulaire. Cette activité se traduit par une stimulation, ou un frein, des voies de signalisation qui gouvernent soit la prolifération, soit la mort cellulaire. Ces processus étant très interdépendants, des effets contradictoires sont parfois observés en fonction du modèle ou du type cellulaire étudié. La sensibilité ou la résistance des cellules cancéreuses à la mort dépend d’un grand nombre de protéines régulatrices clés qui sont cibles du NO (Figure 3).
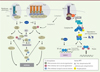 |
Figure 3. S-nitrosylation et voies des récepteurs de mort membranaires de la famille TNFR (TNF receptor). La liaison des ligands TRAIL (tumor-necrosis-factor-related apoptosis inducing ligand), FasL (Fas ligand) et TNFα (tumor necrosis factor alpha) sur leurs récepteurs respectifs DR4/5 (death receptor 4/5), Fas et TNFR1 (tumor necrosis factor receptor 1) induit la formation d’un complexe intracytoplasmique associé au récepteur appelé DISC (death-inducing signaling complex) et complexe I. La formation du DISC conduit à l’activation des caspases (casp) en cascade conduisant à la mort cellulaire par apoptose. La formation du complexe I permet l’activation de la voie classique NF-kB (nuclear factor-kappa B). Suite à la poly-ubiquitination de RIPK1 (receptor-interacting protein kinase 1) par cIAP1/2 (cellular inhibitor of apoptosis1/2), les complexes LUBAC (linear ubiquitin chain assembly complex), TAK1-TAB2/3 (transforming growth factor-β activated kinase [TAK1]/TAK1-binding 2 [TAB2]/TAK1 binding 3 [TAB3]) sont recrutés et participent à d’autres modifications post-traductionelles (MPT) (poly-ubiquitination). L’activation du complexe IKK (inhibitor of NF-kB kinase) via la phosphorylation de sa sous-unité IKKβ (inhibitor of NF-kB kinase beta), permet la phosphorylation puis la dégradation (via une poly-ubiquitination) du répresseur de NF-kB, IkBα (inhibitor of NF-kB alpha). La translocation de NF-kB dans le noyau permet la transcription de gènes cibles impliqués dans la prolifération et la survie cellulaire. Si la voie classique est rendue inactive, un complexe II (qui dérive du complexe I) rejoint une cascade d’activation des caspases et la mort cellulaire par apoptose. Bcl-2 : B-cell lymphoma 2 ; FADD : Fas (TNFRSF6)-associated via death domain ; cFlip : cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein ; Cyt c : cytochrome c ; Apaf-1 : apoptotic peptidase activating factor 1 ; TRADD : tumor necrosis factor receptor type 1-associated death domain protein ; TRAF2/5 (tumor necrosis factor receptor-associated factor2/5). |
S-nitrosylation et résistance à la mort des cellules cancéreuses
Une forte activité de la NOS inductible (iNOS) associée à un taux élevé de NO est fréquemment observée dans les tumeurs et peut être corrélée à une chimiorésistance. L’impact de la S-nitrosylation dans la résistance des cellules cancéreuses à un stimulus apoptotique peut se traduire par une perte de fonction de protéines pro-apoptotiques et/ou une augmentation de l’expression de protéines anti-apoptotiques.
Les caspases sont les acteurs moléculaires clés de la mort cellulaire par apoptose. Elles sont aussi des cibles incontournables du NO. Elles sont un exemple bien caractérisé de l’effet inhibiteur que présente la S-nitrosylation sur leur activité et leur mode d’activation, participant à la résistance des cellules cancéreuses à l’apoptose. Toutes les caspases possèdent au sein de leur site catalytique une cystéine indispensable à leur activité enzymatique, également cible privilégiée du NO. En l’absence de stimulus apoptotique, les caspases sont présentes sous forme de proenzymes inactives. La S-nitrosylation des caspases-8, -9 et -3 retrouvée à l’état basal dans des cellules cancéreuses apparaît donc comme un niveau supplémentaire dans la régulation négative de ces protéines en l’absence de signal pro-apoptotique. L’inhibition de l’activité enzymatique de la caspase-8 par S-nitrosylation protège les hépatocytes de rat de l’apoptose induite par le TNFα (tumor necrosis factor a) [14], tandis que dans des cellules de carcinome colique humain, la S-nitrosylation de la procaspase-9 prévient son clivage et son activation consécutive [15]. Cependant, la stimulation du récepteur apoptotique Fas permet la dénitrosylation de la caspase-3 par la thioredoxine réductase 2 (TrxR2) [16]. Dans ce contexte, il a été démontré plus récemment, que l’IL-24 jouait un rôle primordial, en amont [17]. La transnitrosylation constitue indirectement un autre mécanisme de dénitrosylation des caspases. Ainsi, la transnitrosylation de la caspase-3 vers son inhibiteur endogène XIAP (X-linked inhibitor of apoptosis) induit la S-nitrosylation et l’inactivation de ce dernier, c’est-à-dire son activité anticaspase et, par conséquent, lève l’inhibition de la caspase-3. Si ce mécanisme a pu être mis en évidence dans un contexte de maladie neurodégénérative favorisant l’activité de la caspase-3, il n’a jamais été observé dans des cellules cancéreuses [18, 19].
Bcl-2 (B-cell lymphoma 2) et cFLIP (cellular FLICE [FADD-like IL-1b-converting enzyme]-inhibitory protein) sont des régulateurs négatifs importants des deux principales voies d’induction de l’apoptose, respectivement, la voie mitochondriale et la voie des récepteurs de mort. Sous différentes conditions, leur dégradation favorise le déclenchement de l’apoptose. À l’inverse, leur surexpression est associée à l’échappement à l’apoptose qui est observé lors la carcinogenèse ou encore la chimiorésistance. De fait, la S-nitrosylation de Bcl-2 sur les cystéines Cys158 et Cys229 et de cFLIP sur les cystéines Cys254 et Cys259 augmente leur stabilité en prévenant leur ubiquitination et la dégradation par le protéasome [20, 21]. L’activation du facteur de transcription NF-kB (nuclear factor kappa B) occupe également une place importante dans le phénomène de résistance des cellules à l’apoptose. En effet, sous sa forme S-nitrosylée, cFLIP stimule l’activité de NF-κB aussi bien via la signalisation impliquant le récepteur Fas et son ligand FasL que celle du TNFα et de son récepteur, TNFR1 (tumor necrosis factor-receptor 1) [22, 23]. Sous l’effet du TNFα, cette activation, d’une part, reposerait sur le recrutement et l’adressage de la protéine RIPK1 (receptor-interacting serine/threonine kinase 1) au complexe de signalisation I (TNFR1 signalling complex I), et, d’autre part, produirait un fragment de clivage impliqué dans l’activation de la voie NF-kB [24]. La chimiorésistance de cellules cancéreuses pulmonaires au cisplatine2 est également modulée par le NO. Elle repose sur la S-nitrosylation et la stabilisation de l’expression de Bcl-2 [25]. Des travaux ont d’ailleurs montré que l’IL-24 induisait la dénitrosylation de Bcl-2, en diminuant le taux de NO via une réduction du taux de l’enzyme iNOS et une augmentation de la thioredoxine-1 (TrxR1).
S-nitrosylation et sensibilisation à la mort des cellules cancéreuses
Bien souvent, les voies de signalisation de mort cellulaire engagées par les chimiothérapies et/ou les radiothérapies conventionnelles reposent sur la protéine p53. Cible de multiples mutations retrouvées dans plus de 50 % des cancers, l’inactivation de p53 contribue inévitablement à la chimiorésistance. Aussi, l’activation de voies de signalisation alternatives (indépendantes de p53) telles que celles initiées par les récepteurs de mort membranaires Fas, TRAIL-R1/DR4 ou -R2/DR5 (TNF-related apoptosis-inducing ligand-receptor 1/2 / death receptor 4/5) ou encore TNF-R1, représente une stratégie intéressante pour contrecarrer ce problème majeur dans le traitement des cancers. Plusieurs composantes de la signalisation initiée par les récepteurs du TNF, ciblées par le NO, participent à la sensibilisation des cellules cancéreuses à la mort. Les mécanismes moléculaires associés à cet effet impliquent, entre autres, la S-nitrosylation de leurs récepteurs respectifs. Le nitrosocobalamin (NO-Cbl), un analogue de la vitamine B12, donneur de NO, sensibilise les cellules cancéreuses spécifiquement à TRAIL par la S-nitrosylation de son récepteur TRAIL-R1/DR4 [26]. De façon similaire, le glycéryl trinitrate, ou GTN, sensibilise les cellules cancéreuses du côlon et du sein à l’apoptose induite par FasL [27]. Si la compréhension des mécanismes moléculaires exacts de la signalisation induite dans le cas de NO-Cbl/TRAIL demeure incomplète, il est maintenant admis que la S-nitrosylation de Fas sur la cystéine Cys304 facilite son agrégation et sa relocalisation dans les radeaux lipidiques de la membrane plasmique facilitant la transmission du signal [27]. Par ailleurs, la S-nitrosylation de Ying Yang 1 (YY1), un répresseur transcriptionnel de Fas, participe à cet effet. Elle prévient son activité permettant une expression plus importante de Fas.
Plusieurs études montrent que la S-nitrosylation est une MPT qui inhibe la voie TNFα/TNF-R1. Dans les conditions physiologiques normales, et en l’absence d’activation du récepteur TNF-R1, la S-nitrosylation de la sous-unité IKKβ (inhibitor of NF-kB kinase b) sur la cystéine Cys179, au sein du complexe IKK, de même que celle des sous-unités p50 et p65 de l’hétérodimère NF-kB respectivement sur les cystéines Cys62 et Cys38, contribuent à maintenir inactive la voie de signalisation classique NF-kB [28–31]. Sous l’effet du TNFα, ou d’un autre stimulus (comme le lipopolysaccharide bactérien [LPS]), la dénitrosylation de ces protéines peut rétablir l’activation de la voie de signalisation [32]. L’inhibition de la voie classique NF-kB représente une cible thérapeutique pertinente pour le développement de drogues anticancéreuses visant à freiner la progression, voire même promouvoir la régression tumorale. L’effet antitumoral observé dans les cancers coliques de certains donneurs de NO de type NO-NSAID (NO-releasing non-steroidal anti-inflammatory drugs) comme le NO-aspirine (NO-ASA) ou encore le NO-naproxen, repose en partie sur la S-nitrosylation et l’inhibition de NF-kB [33, 34].
Donneur de NO et thérapie anticancéreuse
Études précliniques
Les stratégies thérapeutiques envisagées consistent à délivrer du NO de façon concomitante à une chimiothérapie afin d’en améliorer les effets. Plusieurs études in vitro démontrent et mettent en exergue l’effet sensibilisateur des donneurs de NO à la mort par apoptose de cellules cancéreuses traitées ou non par chimiothérapie ou radiothérapie. Une inhibition de la croissance tumorale a également été constatée dans différents modèles murins de xénogreffes tumorales humaines après traitement par un donneur de NO en association avec une chimiothérapie et/ou radiothérapie. En effet, dans plusieurs types de cancers, une régression tumorale efficace a été obtenue en combinant différents donneurs de NO avec le ciplastine (Tableau I). Un dérivé du NO-aspirine (le NCX-4040) associé au cisplatine a aussi permis de restaurer la sensibilité des tumeurs ovariennes humaines résistantes à cette chimiothérapie [35]. De même, combiné à l’oxaliplatine, le NCX-4040 a montré, in vivo, une activité antitumorale contre les tumeurs coliques humaines résistantes ou non à la doxorubicine3 [36]. Enfin, la trinitrine, ou glycéryl trinitrate (GTN), un autre donneur de NO, sensibilise, in vivo, à la doxorubicine les tumeurs de la prostate humaine [37].
Régression tumorale observée après traitement combinant un donneur de NO et une chimiothérapie et/ou radiothérapie dans des modèles murins de xénogreffe de tumeurs humaines. DETA/NONOate : diéthylène triamine/diazeniumdiolate ; GSNO : S-nitrosoglutathione ; GTN : glycéryl trinitrate ; NF-kB : nuclear factor-kappa beta ; YY1 : ying yang-1 ; Bcl-XL : B-cell lymphoma-extra large ; AIF : apoptosis inducing factor ; STAT3 : signal transducer and activator of transcription 3 ; EGFR : epidermal growth factor receptor ; PI3K : phosphatidylinositol-3 kinase.
Études cliniques
La trinitrine (GTN) est un médicament utilisé depuis longtemps pour le traitement des angines de poitrine. Cet effet, qui est indésirable en thérapie anticancéreuse, est limité par l’acquisition par les patients d’une tolérance à ce traitement. Son utilisation en stratégie anticancéreuse est en pleine investigation. Les premières études cliniques de phase II ont montré le bénéfice thérapeutique qu’apporte le GTN dans la réponse de patients atteints du cancer du poumon et de la prostate [38, 39]. En effet, chez certains patients porteurs d’un cancer du poumon non à petites cellules à un stade avancé, le GTN améliore l’effet d’une chimiothérapie de première ligne (telle que vinorelbine/cisplatine) [38]. Cet essai clinique pivot a conforté la réalisation d’un essai de phase III réalisé chez le même type de patients mais pour lesquels la combinaison du GTN avec le carboplatine n’avait toutefois pas démontré d’effet bénéfique [40]. Un autre essai clinique de phase II, mené également chez ce même type de patients, n’a cependant pas permis de mettre en évidence un effet bénéfique du GTN lorsqu’utilisé en conjonction avec une chimiothérapie de première ligne constituée de carboplatine/paclitaxel/bevacizumab [41]. Le GTN semble néanmoins améliorer le pronostic des patients atteints du cancer de la prostate après radiothérapie [39]. D’autres essais cliniques de phase I et II associant le GTN avec de la chimio-radiothérapie concomitante montrent que la combinaison est bien tolérée [42, 43].
Conclusion
Le besoin fondamental de comprendre les bases moléculaires régulant les principales voies de signalisation impliquées dans les processus de survie et de mort cellulaire est un défi de taille pour le développement de nouvelles stratégies de lutte contre le cancer. L’une des difficultés dans la recherche des mécanismes moléculaires qui impliquent le NO est la complexité due à son double jeu selon le contexte cellulaire dans lequel il agit. À l’instar de nouvelles molécules anticancéreuses qui sont testées en clinique, plusieurs composés participant aux voies de signalisation de survie ou de mort cellulaire et qui sont cibles du NO, exercent une action similaire. Cela permet donc d’envisager les donneurs de NO comme de nouveaux espoirs thérapeutiques. De nombreuses classes de donneurs de NO existent (Tableau II) et les progrès dans leur synthèse ont permis de développer de nouvelles molécules qui sont mieux tolérées par l’organisme, comme les dérivés du NO-aspirine, qui s’avèrent très prometteuses en thérapie anticancéreuse. Une nouvelle génération de donneurs de NO innovants émerge. Ils utilisent des nanoparticules qui permettent d’améliorer leurs activités antitumorales. Les donneurs de NO représentent donc de potentiels agents thérapeutiques. Néanmoins, leur association avec une chimiothérapie, une radiothérapie ou encore une immunothérapie, nécessite de plus amples essais cliniques afin de valider leur utilisation.
Les principales classes de donneurs de NO. *Donneur de NO faisant ou ayant fait l’objet d’un essai clinique.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
État rédox : indique le pouvoir oxydant ou réducteur d’une substance.
Les sels de platine sont des agents proches des alkylants. Ils forment des liens chimiques entre les différents brins d’ADN et à l’intérieur d’un brin lui-même.
La doxorubicine (Adriamycine™) se fixe sur les structures nucléaires de la cellule, bloquant la synthèse de l’ADN et de l’ARN : c’est un intercalant de l’ADN.
Références
- Murillo-Carretero M, Torroglosa A, Castro C, et al. S-Nitrosylation of the epidermal growth factor receptor: a regulatory mechanism of receptor tyrosine kinase activity. Free Radic Biol Med 2009 ; 46 : 471–479. [CrossRef] [PubMed] [Google Scholar]
- Lam YW, Yuan Y, Isaac J, et al. Comprehensive identification and modified-site mapping of S-nitrosylated targets in prostate epithelial cells. PLoS One 2010 ; 5 : e9075. [CrossRef] [PubMed] [Google Scholar]
- Switzer CH, Glynn SA, Cheng RY, et al. S-nitrosylation of EGFR and Src activates an oncogenic signaling network in human basal-like breast cancer. Mol Cancer Res 2012 ; 10 : 1203–1215. [CrossRef] [PubMed] [Google Scholar]
- Switzer CH, Cheng RY, Ridnour LA, et al. Ets-1 is a transcriptional mediator of oncogenic nitric oxide signaling in estrogen receptor-negative breast cancer. Breast Cancer Res 2012 ; 14 : R125. [CrossRef] [PubMed] [Google Scholar]
- Feng X, Sun T, Bei Y, et al. S-nitrosylation of ERK inhibits ERK phosphorylation and induces apoptosis. Sci Rep 2013 ; 3 : 1814. [PubMed] [Google Scholar]
- Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for cancer treatment. Annu Rev Med 2016 ; 67 : 11–28. [CrossRef] [PubMed] [Google Scholar]
- Lopez-Rivera E, Jayaraman P, Parikh F, et al. Inducible nitric oxide synthase drives mTOR pathway activation and proliferation of human melanoma by reversible nitrosylation of TSC2. Cancer Res 2014 ; 74 : 1067–1078. [CrossRef] [Google Scholar]
- Yu CX, Li S, Whorton AR. Redox regulation of PTEN by S-nitrosothiols. Mol Pharmacol 2005 ; 68 : 847–854. [PubMed] [Google Scholar]
- Yasukawa T, Tokunaga E, Ota H, et al. S-nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. J Biol Chem 2005 ; 280 : 7511–7518. [CrossRef] [PubMed] [Google Scholar]
- Asanuma K, Huo X, Agoston A, et al. In oesophageal squamous cells, nitric oxide causes S-nitrosylation of Akt and blocks SOX2 (sex determining region Y-box 2) expression. Gut 2015 ; doi : 10.1136/gutjnl-2015-309272 [Google Scholar]
- Nath N, Vassell R, Chattopadhyay M, et al. Nitro-aspirin inhibits MCF-7 breast cancer cell growth: effects on COX-2 expression and Wnt/beta-catenin/TCF-4 signaling. Biochem Pharmacol 2009 ; 78 : 1298–1304. [CrossRef] [PubMed] [Google Scholar]
- Nath N, Kashfi K, Chen J, Rigas B. Nitric oxide-donating aspirin inhibits beta-catenin/T cell factor (TCF) signaling in SW480 colon cancer cells by disrupting the nuclear beta-catenin-TCF association. Proc Natl Acad Sci USA 2003 ; 100 : 12584–12589. [CrossRef] [Google Scholar]
- Razavi R, Gehrke I, Gandhirajan RK, et al. Nitric oxide-donating acetylsalicylic acid induces apoptosis in chronic lymphocytic leukemia cells and shows strong antitumor efficacy in vivo. Clin Cancer Res 2011 ; 17 : 286–293. [CrossRef] [PubMed] [Google Scholar]
- Kim YM, Kim TH, Chung HT, et al. Nitric oxide prevents tumor necrosis factor alpha-induced rat hepatocyte apoptosis by the interruption of mitochondrial apoptotic signaling through S-nitrosylation of caspase-8. Hepatology 2000 ; 32 : 770–778. [CrossRef] [PubMed] [Google Scholar]
- Kim JE, Tannenbaum SR. S-Nitrosation regulates the activation of endogenous procaspase-9 in HT-29 human colon carcinoma cells. J Biol Chem 2004 ; 279 : 9758–9764. [CrossRef] [PubMed] [Google Scholar]
- Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science 2008 ; 320 : 1050–1054. [CrossRef] [PubMed] [Google Scholar]
- Tian H, Zhang DF, Zhang BF, et al. Melanoma differentiation associated gene-7/interleukin-24 induces caspase-3 denitrosylation to facilitate the activation of cancer cell apoptosis. J Interferon Cytokine Res 2015 ; 35 : 157–167. [CrossRef] [PubMed] [Google Scholar]
- Tsang AH, Lee YI, Ko HS, et al. S-nitrosylation of XIAP compromises neuronal survival in Parkinson’s disease. Proc Natl Acad Sci USA 2009 ; 106 : 4900–4905. [CrossRef] [Google Scholar]
- Nakamura T, Wang L, Wong CC, et al. Transnitrosylation of XIAP regulates caspase-dependent neuronal cell death. Mol Cell 2010 ; 39 : 184–195. [CrossRef] [PubMed] [Google Scholar]
- Azad N, Vallyathan V, Wang L, et al. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel anti-apoptotic mechanism that suppresses apoptosis. J Biol Chem 2006 ; 81 : 34124–34134. [CrossRef] [Google Scholar]
- Chanvorachote P, Nimmannit U, Wang L, et al. Nitric oxide negatively regulates Fas CD95-induced apoptosis through inhibition of ubiquitin-proteasome-mediated degradation of FLICE inhibitory protein. J Biol Chem 2005 ; 280 : 42044–42050. [CrossRef] [PubMed] [Google Scholar]
- Wang L, Azad N, Kongkaneramit L, et al. The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. J Immunol 2008 ; 180 : 3072–3080. [CrossRef] [PubMed] [Google Scholar]
- Iyer AK, Azad N, Talbot S, et al. Antioxidant c-FLIP inhibits Fas ligand-induced NF-kappaB activation in a phosphatidylinositol 3-kinase/Akt-dependent manner. J Immunol 2011 ; 187 : 3256–3266. [CrossRef] [PubMed] [Google Scholar]
- Talbott SJ, Luanpitpong S, Stehlik C, et al. S-nitrosylation of FLICE inhibitory protein determines its interaction with RIP1 and activation of NF-κB. Cell Cycle 2014 ; 13 : 1948–1957. [CrossRef] [PubMed] [Google Scholar]
- Chanvorachote P, Nimmannit U, Stehlik C, et al. Nitric oxide regulates cell sensitivity to cisplatin-induced apoptosis through S-nitrosylation and inhibition of Bcl-2 ubiquitination. Cancer Res 2006 ; 66 : 6353–6360. [CrossRef] [Google Scholar]
- Tang Z, Bauer JA, Morrison B, Lindner DJ. Nitrosylcobalamin promotes cell death via S nitrosylation of Apo2L/TRAIL receptor DR4. Mol Cell Biol 2006 ; 26 : 5588–5594. [CrossRef] [PubMed] [Google Scholar]
- Leon-Bollotte L, Subramaniam S, Cauvard O, et al. S-nitrosylation of the death receptor fas promotes fas ligand-mediated apoptosis in cancer cells. Gastroenterology 2011 ; 140 : 2009–2018. [CrossRef] [PubMed] [Google Scholar]
- Colasanti M, Persichini T. Nitric oxide: an inhibitor of NF-kappaB/Rel system in glial cells. Brain Res Bull 2000 ; 52 : 155–161. [CrossRef] [PubMed] [Google Scholar]
- Marshall HE, Stamler JS. Inhibition of NF-kappa B by S-nitrosylation. Biochemistry 2001 ; 40 : 1688–1693. [CrossRef] [PubMed] [Google Scholar]
- Kelleher ZT, Matsumoto A, Stamler JS, Marshall HE. NOS2 regulation of NF-kappaB by S-nitrosylation of p65. J Biol Chem 2007 ; 282 : 30667–30672. [CrossRef] [PubMed] [Google Scholar]
- Reynaert NL, Ckless K, Korn SH, et al. Nitric oxide represses inhibitory kappaB kinase through S-nitrosylation. Proc Natl Acad Sci USA 2004 ; 101 : 8945–8950. [CrossRef] [Google Scholar]
- Kelleher ZT, Potts EN, Brahmajothi MV, et al. NOS2 regulation of LPS-induced airway inflammation via S-nitrosylation of NF-kB p65. Am J Physiol Lung Cell Mol Physiol 2011 ; 301 : L327–L333. [CrossRef] [PubMed] [Google Scholar]
- Chattopadhyay M, Goswami S, Rodes DB, et al. NO-releasing NSAIDs suppress NF-κB signaling in vitro and in vivo through S-nitrosylation. Cancer Lett 2010 ; 298 : 204–211. [CrossRef] [PubMed] [Google Scholar]
- Williams JL, Ji P, Ouyang N, et al. Protein nitration and nitrosylation by NO-donating aspirin in colon cancer cells: Relevance to its mechanism of action. Exp Cell Res 2011 ; 317 : 1359–1367. [CrossRef] [PubMed] [Google Scholar]
- Bratasz A, Selvendiran K, Wasowicz T, et al. NCX-4040, a nitric oxide-releasing aspirin, sensitizes drug-resistant human ovarian xenograft tumors to cisplatin by depletion of cellular thiols. J Transl Med 2008 ; 6 : 9. [CrossRef] [PubMed] [Google Scholar]
- Tesei A, Zoli W, Fabbri F, et al. NCX 4040, an NO-donating acetylsalicylic acid derivative: efficacy and mechanisms of action in cancer cells. Nitric Oxide 2008 ; 19 : 225–236. [CrossRef] [PubMed] [Google Scholar]
- Frederiksen LJ, Sullivan R, Maxwell LR, et al. Chemosensitization of cancer in vitro and in vivo by nitric oxide signaling. Clin Cancer Res 2007 ; 13 : 2199–2206. [CrossRef] [PubMed] [Google Scholar]
- Yasuda H, Yamaya M, Nakayama K, et al. Randomized phase II trial comparing nitroglycerin plus vinorelbine and cisplatin with vinorelbine and cisplatin alone in previously untreated stage IIIB/IV non-small-cell lung cancer. J Clin Oncol 2006 ; 24 : 688–694. [CrossRef] [PubMed] [Google Scholar]
- Siemens DR, Heaton JP, Adams MA, et al. Phase II study of nitric oxide donor for men with increasing prostate-specific antigen level after surgery or radiotherapy for prostate cancer. Urology 2009 ; 74 : 878–883. [CrossRef] [PubMed] [Google Scholar]
- Davidson A, Veillard AS, Tognela A, et al. A phase III randomized trial of adding topical nitroglycerin to first-line chemotherapy for advanced nonsmall-cell lung cancer: the Australasian lung cancer trials group NITRO trial. Ann Oncol 2015 ; 26 : 2280–2286. [CrossRef] [PubMed] [Google Scholar]
- Dingemans AM, Groen HJ, Herder GJ, et al. A randomized phase II study comparing paclitaxel-carboplatin-bevacizumab with or without nitroglycerin patches in patients with stage IV nonsquamous nonsmall-cell lung cancer: NVALT12 (NCT01171170). Ann Oncol 2015 ; 26 : 2286–2293. [CrossRef] [PubMed] [Google Scholar]
- Illum H, Wang DH, Dowell JE, et al. Phase I dose escalation trial of nitroglycerin in addition to 5-fluorouracil and radiation therapy for neoadjuvant treatment of operable rectal cancer. Surgery 2015 ; 158 : 460–465. [CrossRef] [PubMed] [Google Scholar]
- Arrieta O, Blake M, de la Mata-Moya MD, et al. Phase II study. Concurrent chemotherapy and radiotherapy with nitroglycerin in locally advanced non-small cell lung cancer. Radiother Oncol 2014 ; 111 : 311–315. [CrossRef] [PubMed] [Google Scholar]
- Huerta-Yepez S, Baritaki S, Baay-Guzman G, et al. Contribution of either YY1 or BclXL-induced inhibition by the NO-donor DETANONOate in the reversal of drug resistance, both in vitro and in vivo. YY1 and BclXL are overexpressed in prostate cancer. Nitric Oxide 2013 ; 29 : 17–24. [CrossRef] [PubMed] [Google Scholar]
- Huerta S, Baay-Guzman G, Gonzalez-Bonilla CR, et al. In vitro and in vivo sensitization of SW620 metastatic colon cancer cells to CDDP-induced apoptosis by the nitric oxide donor DETANONOate: Involvement of AIF. Nitric Oxide 2009 ; 20 : 182–194. [CrossRef] [PubMed] [Google Scholar]
- Gao X, Saha D, Kapur P, et al. Radiosensitization of HT-29 cells and xenografts by the nitric oxide donor DETANONOate. J Surg Oncol 2009 ; 100 : 149–158. [CrossRef] [PubMed] [Google Scholar]
- Kaliyaperumal K, Sharma AK, McDonald DG, et al. S-nitrosoglutathione-mediated STAT3 regulation in efficacy of radiotherapy and cisplatin therapy in head and neck squamous cell carcinoma. Redox Biol 2015 ; 6 : 41–50. [CrossRef] [PubMed] [Google Scholar]
- Selvendiran K, Bratasz A, Tong L, et al. NCX-4016, a nitro-derivative of aspirin, inhibits EGFR and STAT3 signaling and modulates Bcl-2 proteins in cisplatin-resistant human ovarian cancer cells and xenografts. Cell Cycle 2008 ; 7 : 81–88. [CrossRef] [PubMed] [Google Scholar]
- Hess DT, Stamler JS. Regulation by S-nitrosylation of protein post-translational modification. J Biol Chem 2012 ; 287 : 4411–4418. [CrossRef] [PubMed] [Google Scholar]
Liste des tableaux
Régression tumorale observée après traitement combinant un donneur de NO et une chimiothérapie et/ou radiothérapie dans des modèles murins de xénogreffe de tumeurs humaines. DETA/NONOate : diéthylène triamine/diazeniumdiolate ; GSNO : S-nitrosoglutathione ; GTN : glycéryl trinitrate ; NF-kB : nuclear factor-kappa beta ; YY1 : ying yang-1 ; Bcl-XL : B-cell lymphoma-extra large ; AIF : apoptosis inducing factor ; STAT3 : signal transducer and activator of transcription 3 ; EGFR : epidermal growth factor receptor ; PI3K : phosphatidylinositol-3 kinase.
Les principales classes de donneurs de NO. *Donneur de NO faisant ou ayant fait l’objet d’un essai clinique.
Liste des figures
|
Figure 1. La cystéine au cœur de la S-nitrosylation. S-nitrosylation : régulation de l’activité des protéines. De nombreuses enzymes impliquées dans la MPT (modification post-traductionelle) des protéines possèdent un résidu cystéine dans leur site catalytique. La réactivité du monoxyde d’azote (NO) avec un tel résidu abroge la plupart du temps leur activité. Les enzymes de types kinases (par exemple IKKβ [inhibitor of NF-kB kinase beta]), phosphatases (par exemple PTEN [phosphatase and tensin homolog]), E3 ubiquitine ligases (par exemple XIAP [X-linked inhibitor of apoptosis]) sont souvent la cible du NO au sein de différentes voies de signalisation contrôlant la survie ou la mort cellulaire. De ce fait, la S-nitrosylation d’une cystéine spécifique peut influencer la survenue d’un grand nombre de MPT et par conséquent altérer plusieurs voies de signalisation cellulaire aussi bien positivement que négativement. S-nitrosylation : régulation de la structure et stabilité des protéines. Les groupements thiols formant ensemble des ponts disulfures ne réagissent pas avec le NO et par conséquent ne constituent pas des sites de S-nitrosylation. Néanmoins deux sites S-nitrosylés peuvent a posteriori réagir entre eux pour former un pont disulfure (par exemple Akt1 [RAC-alpha serine/threonine-protein kinase]) contribuant ainsi à la stabilité de la structure ternaire et quaternaire de la protéine. En revanche, la S-nitrosylation peut perturber la stabilité d’une métalloprotéine qui met en jeu des résidus cystéines ayant des propriétés de liaison avec les métaux. La S-nitrosylation du facteur HIF1α (hypoxia inducible factor 1 alpha) entraîne sa stabilisation. S-nitrosylation : régulation de la fonction et localisation des protéines. La S-nitrosylation peut moduler le rôle d’une protéine lorsque les résidus cystéine ne sont impliqués ni dans le domaine catalytique d’une enzyme ni dans sa stabilité. La S-nitrosylation peut influencer la capacité d’une protéine à interagir avec un partenaire déterminé et contrecarrer sa fonction. Dans le cas de TSC (tuberous sclerosis complex)2, sa S-nitrosylation prévient son interaction avec son partenaire protéique TSC1 et ainsi sa fonction. Au contraire, la S-nitrosylation de cFLIP (cellular FLICE [FADD-like IL-1β-converting enzyme]-inhibitory protein) induit son interaction avec RIPK1 (receptor-interacting protein kinase 1) pour promouvoir sa capacité à induire la voie classique NF-kB (nuclear factor-kappa B) et/ou sa localisation subcellulaire en affectant par exemple les processus de transcription (la protéine GAPDH [glyceraldehyde-3-phosphate dehydrogenase] sous sa forme S-nitrosylée assure son interaction avec la protéine Siah1 [siah E3 ubiquitin protein ligase 1] qui induit sa translocation dans le noyau), de traduction, de dégradation des protéines ou encore de modifications post-traductionnelles. S-nitrosylation : sources de nouvelles MPT. Les propriétés du groupement thiol d’une cystéine en font un site privilégié pour de nombreuses autres MPT telles que la S-palmitoylation (par exemple Ras), la S-glutathionylation (par exemple caspase-3) ou encore la S-sulfhydration (par exemple GAPDH) [49]. Il est important de noter que la S-nitrosylation d’un site peut à la fois prévenir la formation d’une autre MPT ou au contraire servir d’intermédiaire pour la formation d’une nouvelle MPT oxidative selon l’environnement rédox dans lequel il se trouve. |
| Dans le texte | |
|
Figure 2. S-nitrosylation et principales voies de signalisation impliquées dans la prolifération cellulaire. La S-nitrosylation module l’activité (inhibition ou activation) de nombreux acteurs des voies de signalisation MAPK (mitogen-activated protein kinase) et PI3K/AKT (phosphatidylinositol-3-kinase/protein kinase B) initiées via les récepteurs à tyrosine kinase. La voie mTOR (mammalian target of rapamycin) joue un rôle clé dans l’équilibre entre survie et mort cellulaire. Il existe deux complexes protéiques fonctionnellement distincts formés par mTOR : mTORC1 (mammalian target of rapamycin complex 1) (associé entre autres à raptor) et mTORC2 (mammalian target of rapamycin complex 2) (associé entre autres à rictor). L’activation de la voie Wnt/β-caténine (wingless and int-1/beta-catenin) déstabilise le complexe de destruction de la β-caténine, APC (adenomatous polyposis coli), Axine, CKIα (casein kinase I a), GSK3β (glycogen synthase kinase 3). La S-nitrosylation de la β-caténine prévient non seulement son interaction avec le facteur de transcription TCF-4 (T cell transcription factor 4) mais sert aussi de signal précédant sa dégradation par le protéasome. L’ensemble des flèches correspond à la transduction du signal lorsque les voies sont normalement actives. MEK : mitogen-activated protein kinase kinase (ou MAP2K) ; ERK : extracellular signal-regulated kinases ; PKC : protéine kinase C ; SGK : serum-and glucocorticoid-induced protein kinase ; TSC : tuberous sclerosis complex ; Rheb : Ras homology enriched in brain ; PTEN : phosphatase and tensin homolog ; PIP2 : phosphatidylinositol-4,5-bisphosphate ; PIP3 : phosphatidylinositol (3,4,5)-trisphosphate ; Dsh : dishevelled ; LRP5/6 : low-density-lipoprotein receptor-related protein 5/6 ; S6K1 : S6 kinase 1 ; 4E-BP1 : 4E-binding protein 1 ; Ets-1 : E26 transformation-specific sequence-1. |
| Dans le texte | |
|
Figure 3. S-nitrosylation et voies des récepteurs de mort membranaires de la famille TNFR (TNF receptor). La liaison des ligands TRAIL (tumor-necrosis-factor-related apoptosis inducing ligand), FasL (Fas ligand) et TNFα (tumor necrosis factor alpha) sur leurs récepteurs respectifs DR4/5 (death receptor 4/5), Fas et TNFR1 (tumor necrosis factor receptor 1) induit la formation d’un complexe intracytoplasmique associé au récepteur appelé DISC (death-inducing signaling complex) et complexe I. La formation du DISC conduit à l’activation des caspases (casp) en cascade conduisant à la mort cellulaire par apoptose. La formation du complexe I permet l’activation de la voie classique NF-kB (nuclear factor-kappa B). Suite à la poly-ubiquitination de RIPK1 (receptor-interacting protein kinase 1) par cIAP1/2 (cellular inhibitor of apoptosis1/2), les complexes LUBAC (linear ubiquitin chain assembly complex), TAK1-TAB2/3 (transforming growth factor-β activated kinase [TAK1]/TAK1-binding 2 [TAB2]/TAK1 binding 3 [TAB3]) sont recrutés et participent à d’autres modifications post-traductionelles (MPT) (poly-ubiquitination). L’activation du complexe IKK (inhibitor of NF-kB kinase) via la phosphorylation de sa sous-unité IKKβ (inhibitor of NF-kB kinase beta), permet la phosphorylation puis la dégradation (via une poly-ubiquitination) du répresseur de NF-kB, IkBα (inhibitor of NF-kB alpha). La translocation de NF-kB dans le noyau permet la transcription de gènes cibles impliqués dans la prolifération et la survie cellulaire. Si la voie classique est rendue inactive, un complexe II (qui dérive du complexe I) rejoint une cascade d’activation des caspases et la mort cellulaire par apoptose. Bcl-2 : B-cell lymphoma 2 ; FADD : Fas (TNFRSF6)-associated via death domain ; cFlip : cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein ; Cyt c : cytochrome c ; Apaf-1 : apoptotic peptidase activating factor 1 ; TRADD : tumor necrosis factor receptor type 1-associated death domain protein ; TRAF2/5 (tumor necrosis factor receptor-associated factor2/5). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.