")
")
| Issue |
Med Sci (Paris)
Volume 27, Number 1, Janvier 2011
|
|
|---|---|---|
| Page(s) | 55 - 61 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/201127155 | |
| Published online | 10 février 2011 | |
La fibrose tubulo-interstitielle rénale
Menace fantôme ou dernière croisade ?
Tubulo-interstitial fibrosis
An emerging major health problem
Inserm, Université Toulouse III Paul Sabatier ; Inserm U858, Hôpital Rangueil, 31432 Toulouse, France ; Institut de médecine moléculaire de Rangueil, équipe no. 5, IFR150, Institut Louis Bugnard, avenue Jean Poulhes, Hôpital Rangueil, 31432 Toulouse, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Le vieillissement de la population et l’augmentation du nombre de patients atteints de diabète entraînent une forte accélération de l’incidence des néphropathies menant à l’insuffisance rénale. Quelle qu’en soit l’origine, la majorité des atteintes rénales aboutissent au développement d’une fibrose tubulo-interstitielle (FTI) dont la présence signe l’évolution vers la perte de la fonction rénale. Comprendre et lutter contre le développement de la FTI représente ainsi un enjeu scientifique et médical de la plus grande importance. Dans cette revue, nous analyserons donc quels sont les mécanismes et les facteurs de progression de la FTI et quelles sont les stratégies thérapeutiques envisageables pour l’avenir.
Abstract
The incidence of chronic kidney disease leading to end-stage renal disease has significantly increased and may reach epidemic proportions over the next decade. Regardless of the initial insult, the progression of most forms of renal disease results in tubulo-interstitial fibrosis. This has been closely correlated to the future appearance of renal failure and has therefore been associated with poor long-term prognosis. New molecules and agents to limit the development of tubulo-interstitial fibrosis and slow down the progression towards end-stage renal disease are needed. In the past twenty years, many efforts have been made to understand the mechanisms of tubulo-intersititial fibrosis with the final goal to develop new therapeutic strategies. In this context, this review will focus on the mechanisms and factors involved in the development and the progression of renal fibrosis and will discuss the new promising therapeutic strategies in animal and humans.
© 2011 médecine/sciences – Inserm / SRMS
La fibrose rénale : un problème de santé publique
L’évolution de nos sociétés et de nos modes de vie, l’augmentation de l’incidence de l’obésité, des maladies cardiovasculaires, du diabète, ainsi que le vieillissement de la population entraînent l’accroissement exponentiel du nombre de patients atteints de maladies rénales chroniques et évoluant à plus ou moins long terme vers l’insuffisance rénale terminale. À ce stade, les patients sont soignés par des thérapies de remplacement lourdes et coûteuses comme la dialyse et la transplantation.
Stratégies thérapeutiques : avantages et inconvénients.
Bien que les causes primitives des atteintes rénales soient multiples, la majorité des néphropathies évoluent vers le développement d’une fibrose rénale. La fibrose est définie par une accumulation exagérée de matrice extracellulaire (MEC) [41]. Elle peut se développer dans de nombreux organes (peau, foie, poumons, cœur, rein) et sa présence est souvent associée à la perte de la fonction de l’organe touché. Au niveau rénal, il existe trois grands types de fibrose, dépendant de l’étiologie de la maladie : la fibrose vasculaire, la fibrose glomérulaire (ou glomérulosclérose) et la fibrose tubulo-interstitielle (FTI). À l’heure actuelle, la majorité des maladies rénales chroniques ont pour origine une atteinte glomérulaire et sont donc associées à des lésions de glomérulosclérose. Cependant, la fibrose progresse ensuite presque toujours vers le compartiment tubulo-interstitiel [1] ; d’un point de vue clinique, la présence de FTI est fortement corrélée à une future évolution vers l’insuffisance rénale et est ainsi associée à un mauvais pronostic à long terme [2]. Pour toutes ces raisons, le développement de la FTI, qui fut pendant longtemps une menace silencieuse, est sur le point de devenir un problème majeur de santé publique et ce, au niveau mondial. Au cours des dernières années, de grands efforts de recherche ont ainsi été entrepris afin de mieux comprendre les mécanismes de la FTI et de pouvoir, un jour, développer de nouvelles stratégies thérapeutiques et entrer en guerre contre cette maladie.
La trilogie de la fibrose rénale
La FTI est un processus complexe, qui implique de nombreuses molécules et de nombreux types cellulaires, résidents ou infiltrés. Il est possible de diviser de manière schématique le développement de la FTI en trois phases distinctes : la phase inflammatoire, la phase d’apparition de cellules sécrétrices de matrice extracellulaire et enfin la phase d’accumulation de cette matrice.
Phase inflammatoire : le rôle des macrophages (Dr Jekyll et Mr Hyde)
En réponse à l’atteinte initiale, les cellules rénales résidentes stressées produisent des cytokines pro-inflammatoires et des chimiokines, induisant de ce fait une réponse inflammatoire chronique (Figure 1) [3]. Au niveau rénal, on observe ainsi une augmentation de l’expression des chimiokines et des molécules d’adhésion, associée à une infiltration massive de cellules inflammatoires [3, 4]. D’un point de vue quantitatif aussi bien que qualitatif, les macrophages représentent la population de leucocytes infiltrés la plus importante et la plus fortement impliquée dans l’initiation et la progression de la FTI [3]. Leur degré d’infiltration est d’ailleurs considéré comme étant un facteur prédictif de la progression des néphropathies [5]. Ce concept doit cependant désormais être modulé. En effet, il a été démontré que, dans certaines conditions, en particulier dans les phases plus tardives de la FTI, les macrophages exercent un rôle protecteur. Alors comment expliquer ce rôle contradictoire, à la fois Dr Jekyll et Mr Hyde, des macrophages dans la FTI ? Une fois présents dans le tissu inflammatoire, les monocytes peuvent se différencier en deux familles spécialisées de macrophages (Figure 2) : les macrophages de type M1 (voie classique), apparaissant lorsque les monocytes sont activés par l’IFN- γ (interféron-γ) et le TNF-α (tumor necrosis factor-α), ou les macrophages de type M2 (voie alternative), activés par les interleukines IL-4, IL-13, IL-10, ou encore le TGF-β (transforming growth factor-β) [6]. Les macrophages M1, délétères, produisent des radicaux libres oxygénés et sécrètent des cytokines pro-inflammatoires telles que le TNF-α et l’IL-1-β. Ces produits étant toxiques pour les cellules résidentes rénales, ils contribuent ainsi à l’extension des lésions et à la progression de la fibrose (Figure 2) [6, 7]. À l’inverse, les macrophages de type M2 exercent un rôle protecteur. En effet, bien qu’ils soient capables de produire du TGF-β et de la MEC, phénomènes qui a priori sont associés à un effet pro-fibrosant, leur rôle principal est de réduire l’inflammation grâce à la sécrétion de cytokines anti-inflammatoires comme l’IL-10 et l’IL-6 (Figure 2) [6, 7]. Ainsi Wang et al. ont mis en évidence que le transfert de macrophages polarisés ex vivo vers un phénotype M2 permettait de réduire l’inflammation et le développement de la fibrose rénale chez l’animal alors que le transfert de macrophages de type M1 aggravait les lésions [7]. Bien que les macrophages soient toujours reconnus comme étant les acteurs-clés de l’inflammation associée à la FTI, il est donc désormais nécessaire de mieux caractériser le rôle relatif de ces deux populations. Ceci permettra éventuellement d’envisager des stratégies thérapeutiques dirigées contre l’une de ces deux sous-populations.
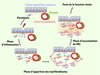 |
Figure 1 Les trois étapes de la FTI. Phase inflammatoire : en réponse à un stress, les cellules rénales résidentes sont activées et produisent des cytokines pro-inflammatoires et des chimiokines. Ceci induit le recrutement et l’infiltration de cellules inflammatoires qui produisent alors des radicaux libres oxygénés ou des cytokines pro-inflammatoires et profibrosantes. Phase d’apparition des myofibroblastes : le contexte inflammatoire mène à l’apparition des myofibroblastes par quatre voies principales : (a) à partir des cellules épithéliales tubulaires (TEM) ; (b) à partir des fibroblastes interstitiels ; (c) à partir des cellules endothéliales des capillaires (TEndM) et (d) à partir des péricytes. Phase d’accumulation de MEC : les myofibroblastes sont les cellules à l’origine de l’accumulation de MEC car ils synthétisent en excès les protéines matricielles mais ils sécrètent également les inhibiteurs des protéases dégradant la MEC. La présence de la FTI signe alors l’évolution vers la perte de la fonction rénale. |
 |
Figure 2 Rôle des macrophages M1 et M2 dans la FTI. Le rôle duel des deux populations distinctes de macrophages est un phénomène reconnu dans le développement de la fibrose rénale. Ainsi, la stimulation chronique des macrophages de type M1 contribue à l’extension des lésions et au développement de la fibrose alors que les macrophages M2 permettent la diminution de l’inflammation, le remodelage matriciel et la régénération tissulaire. IFN : interféron ; IL- : interleukine ; TGF-β : transforming growth factor-β ; MEC : matrice extracellulaire ; TNF- α : tumor necrosis factor. |
Enfin, il est important de noter que parallèlement aux macrophages, les cellules dendritiques [8, 9], les mastocytes [10] et les lymphocytes B et T [4] s’accumulent également dans le rein lors de l’inflammation. Néanmoins, à l’heure actuelle, il n’existe que peu de données fonctionnelles sur le rôle de ces cellules dans le développement de la FTI.
Phase d’apparition des myofibroblastes : un pour tous, tous pour un
Le développement d’un tel contexte inflammatoire représente un stress majeur pour les cellules résidentes rénales et a pour conséquence l’apparition d’un nouveau type cellulaire : les myofibroblastes (Figure 1). Le premier réservoir de myofibroblastes est constitué par les fibroblastes interstitiels résidents (Figure 1). Dans le rein sain, ces fibroblastes sont peu nombreux. Ce sont des cellules mésenchymateuses quiescentes, servant principalement à maintenir l’architecture et la trophicité du rein. En revanche, au cours de la phase inflammatoire, les macrophages activés, aidés des cellules rénales résidentes lésées, synthétisent de grandes quantités de TGF-β et de CTGF (connective tissue growth factor), deux cytokines capables de stimuler la prolifération et l’activation des fibroblastes en myofibroblastes [11]. Même si le débat reste toujours ouvert, il a été montré que d’autres types cellulaires pouvaient également contribuer à l’apparition des myofibroblastes : en réponse au TGF-β et au CTGF, les cellules épithéliales tubulaires ou les cellules endothéliales des vaisseaux peuvent s’engager dans une transition phénotypique au cours de laquelle elles perdent leur phénotype initial, se transforment en cellules mésenchymateuses et envahissent l’interstitium rénal, participant ainsi à l’accumulation des myofibroblastes (Figure 1) [11]. Ces processus sont connus respectivement sous les noms de transition épithélio-mésenchymateuse (TEM) et de transition endothélio-mésenchymateuse (TEndM). Par ailleurs, de plus en plus d’études suggèrent que les péricytes, cellules constitutives de la paroi des vaissaux, représenteraient un important réservoir de myofibroblastes au cours de la FTI (Figure 1) [11].
Phase d’accumulation de MEC
Les myofibroblastes sont les principales cellules responsables de l’accumulation de MEC [3, 5]. D’une part, sous l’action du TGF-β et du CTGF, ces cellules vont synthétiser de grandes quantités de protéines matricielles, telles que la fibronectine ou les collagènes de types I et III. D’autre part, elles vont inhiber la dégradation de ces protéines en synthétisant des inhibiteurs de protéases tels que PAI-1 (plasminogen activator inhibitor-1) ou les TIMP (tissue inhibitor of metalloprotease) (Figure 1) [5]. Parallèlement à leur accumulation, les protéines matricielles sont également modifiées et réticulées par la transglutaminase tissulaire Tg-2, ce qui les rend particulièrement résistantes à la dégradation par les protéases.
Ainsi, lors de la fibrose, les cytokines profibrosantes synthétisées par les macrophages infiltrés et les cellules rénales lésées induisent l’activation et la différenciation de multiples types cellulaires, ces phénomènes convergeant tous vers l’apparition des myofibroblastes et l’accumulation de MEC. De plus, les myofibroblastes synthétisent eux-mêmes de grandes quantités de TGF-β et de CTGF, faisant ainsi basculer le processus dans un cercle vicieux à l’origine de la propagation et de l’aggravation des lésions.
Mécanismes de progression de la fibrose : un tableau toujours plus complexe
Chimiokines, cytokines, facteurs de croissance
Parallèlement à l’effet chimiotactique qui les définit, les chimiokines possèdent également d’autres activités biologiques. Ainsi, la chimiokine CCL2 exerce des effets profibrosants directs, en stimulant la production de TGF-β par les macrophages [12] et la synthèse de collagène et de fibronectine par des cellules rénales [13]. Par conséquent, non seulement les chimiokines déclenchent la phase inflammatoire en stimulant le recrutement des leucocytes au site de la lésion, mais elles participent également activement à l’entretien et à la progression de la fibrose (Figure 3).
 |
Figure 3 Les mécanismes moléculaires de la progression de la fibrose rénale : un puzzle toujours plus complexe. Il s’agit ici d’avoir une vision un peu plus globale des principaux facteurs qui accélèrent (rouge) ou ou au contraire ralentissent (vert) la progression de la FTI. On voit ici à quel point la fibrose est un processus complexe et redondant. CTGF : connective tissue growth factor ; PDGF : platelet-derived growth factor ; FGF : fibroblast growth factor ; BMP-7 : bone morphogenetic protein -7 ; Ang II : angiotensine II ; LPA : acide lysophosphatidique ; RB1, RB2 : récepteurs des kinines ; HGF : hepatocyte growth factor ; EGF-R : récepteur de l’epidermal growth factor. |
En plus des chimiokines, d’autres cytokines et facteurs de croissance sont impliqués dans la régulation de la progression de la fibrose, positivement ou négativement. Les facteurs de croissance aggravants les plus connus sont bien sûr le TGF-β et le CTGF, mais aussi le PDGF (platelet derived growth factor), le bFGF (basic fibroblast growth factor, FGF-2) et les membres de la famille de l’EGF (epithelial growth factor). In vitro, le PDGF stimule la prolifération et la production de chimiokines par les fibroblastes rénaux [14] et in vivo, son blocage réduit l’inflammation et le développement de la fibrose ( Figure 3 ) [14]. Le FGF-2, quant à lui, stimule la TEM et la prolifération des fibroblastes ( Figure 3) [15] et son expression est augmentée dans des biopsies rénales de patients atteints de différentes néphropathies associées à la fibrose. Enfin, la stimulation de l’EGF-R par l’EGF potentialise l’effet du TGF-β sur la TEM et son blocage in vivo diminue le développement de la FTI dans différents modèles animaux de néphropathies (Figure 3) [16, 42].
À l’inverse, il existe des inhibiteurs endogènes de la fibrose et c’est la balance entre les facteurs pro- et anti-fibrosants qui détermine le devenir et la progression de la fibrose rénale. Les facteurs les plus connus limitant la progression de la FTI sont l’HGF (hepatocyte growth factor) et le BMP-7 (bone morphogenic protein-7). L’HGF exerce d’une part des effets antifibrosants en bloquant la signalisation induite par le TGF-β [17-19] et d’autre part, des effets anti-inflammatoires en inhibant l’expression des chimiokines et des cytokines pro-inflammatoires par les cellules épithéliales tubulaires [17, 19, 20] (Figure 3). Le BMP-7, lui, inhibe l’expression des chimiokines pro-inflammatoires par les cellules épithéliales et entraîne l’inversion de la TEM (Figure 3) [21, 22].
Système rénine-angiotensine (SRA)
Le SRA est un système hypertenseur dont l’angiotensine II (AngII) est le peptide le plus actif. Classiquement décrite comme un agent vasoactif, l’AngII est un peptide multifonctionnel qui exerce ses effets par la stimulation de deux récepteurs à sept domaines transmembranaires, les récepteurs AT1R et AT2R. Il est cependant couramment admis que l’AT1R relaie la plupart des effets classiques de l’AngII, et l’activation de l’AT1R par l’AngII est désormais considérée comme ayant un rôle-clé dans la progression de la FTI. En effet, l’AngII joue sur tous les tableaux de la fibrose (Figure 3) : elle participe directement à l’inflammation et au chimiotactisme en stimulant l’expression des molécules d’adhésion et la synthèse de chimiokines [23-25] ; elle exacerbe l’apparition des myofibroblastes en stimulant la TEM ainsi que la prolifération et la différenciation fibroblastiques [23-26] ; enfin, elle joue un rôle important dans l’accumulation de MEC en stimulant la synthèse de collagène I et de fibronectine par les myofibroblastes mais également en induisant l’expression de PAI-1 et de TIMP-1 [23, 25].
Système kinine-kallikréine (SKK)
Les kinines sont des médiateurs impliqués dans de nombreux processus physiopathologiques et qui agissent par l’activation de deux récepteurs, les récepteurs B1 (RB1) et B2 (RB2) [27]. En activant le RB2, les kinines limitent le développement de la FTI car elles stimulent l’activité des protéases dégradant la MEC (Figure 3) [28]. À l’inverse, via l’activation du RB1, elles stimulent la progression de la FTI en favorisant l’expression des chimiokines par les cellules rénales lésées et le recrutement macrophagique (Figure 3) [29, 30].
Acide lysophosphatidique (LPA)
Parmi tous les acteurs moléculaires impliqués dans la fibrose rénale, le LPA est un peu singulier. En effet, il ne s’agit pas d’une protéine ou d’un peptide, mais d’un phospholipide. Des études réalisées chez des patients atteints de maladies rénales chroniques ont démontré une augmentation de la concentration plasmatique de ce phospholipide [31]. De plus, il a été montré que le LPA induisait la synthèse et la sécrétion de CTGF par les fibroblastes interstitiels et les cellules épithéliales tubulaires in vitro (Figure 3) [31, 32] et que le blocage de son action in vivo réduisait fortement la progression de la FTI [32].
Toxicité de la protéinurie
La présence de protéines dans les urines est une situation anormale résultant d’une atteinte de la barrière de filtration glomérulaire qui devient perméable et laisse alors fuir les macromolécules. Cette protéinurie est un facteur de risque important dans la progression de la FTI et de l’évolution vers l’insuffisance rénale (Figure 3) [33, 34]. En effet, en cas d’excès de protéines dans les urines, les capacités d’endocytose des cellules épithéliales proximales, chargées de recapter les protéines, sont dépassées et les lysosomes sont surchargés. Ceci entraîne une rupture des lysosomes, contribuant ainsi à la progression des lésions. Cette toxicité est définie comme une toxicité non spécifique. Mais certains composés peuvent également contribuer de manière spécifique à la progression de la FTI, comme l’albumine, certaines cytokines et certains facteurs de croissance (dont le TGF-β) ou bien les facteurs du complément, qui une fois passés dans l’urine, vont se retrouver de manière anormale directement en contact avec les cellules épithéliales tubulaires.
Hypoxie chronique : un facteur de progression de la FTI
Depuis 1998, l’hypoxie chronique est considérée comme un facteur de progression de la fibrose rénale. En effet, lorsque l’on examine des biopsies de patients atteints de maladies rénales chroniques, on observe une raréfaction des capillaires qui entourent les tubules, engendrant ainsi une hypoxie tissulaire [35]. Or des études menées in vivo et in vitro ont démontré que l’hypoxie pourrait être une cause directe de la progression de la fibrose [35, 36], en stimulant la production de TGF-β et de CTGF, en induisant la TEM et la prolifération/différenciation des fibroblastes et en stimulant l’accumulation de MEC (Figure 3).
En conclusion, les mécanismes de progression de la fibrose rénale sont multiples. Ainsi la recherche de nouveaux médicaments est un défi majeur car la redondance des voies limite l’impact éventuel du blocage d’un seul de ces acteurs. Néanmoins, la multiplication des nouvelles cibles permet d’élargir l’étendue des possibilités thérapeutiques.
Stratégies thérapeutiques et nouvelles cibles
À l’heure actuelle, les seuls traitements efficaces chez l’homme qui peuvent limiter significativement l’évolution vers l’insuffisance rénale sont ceux qui inhibent soit la production d’AngII (IEC, inhibiteurs de l’enzyme de conversion), soit l’action de l’AngII sur son récepteur AT1R (ARA2, antagonistes du récepteur de type 1 à l’AngII) (Tableau I). Malgré ces résultats prometteurs, ces traitements ne font que ralentir la progression de la FTI. Il est donc désormais nécessaire de développer de nouvelles stratégies thérapeutiques qui, utilisées seules ou en combinaison, pourraient stopper, voire inverser la progression de la FTI et l’évolution vers l’insuffisance rénale (voir Encadré).
Réversibilité de la FTI : une réalité chez la souris
En 2006, le caractère irrévocable de la FTI chez l’homme a été remis en question par une étude clinique de Fioretto et al. quand les auteurs ont mis en évidence qu’il était possible d’obtenir une réversibilité des lésions de FTI chez des patients atteints d’un diabète de type 1 après transplantation du pancréas [37]. Pour autant, ces résultats restent encore extrêmement anecdotiques. Paradoxalement chez l’animal, un grand nombre d’études ont permis de mettre en évidence le phénomène de réversibilité [38]. Par conséquent, pourquoi les traitements marchent-ils si bien chez l’animal et si peu chez l’homme ? Tout d’abord, il est évidemment plus facile d’administrer des doses plus fortes, et donc potentiellement plus efficaces, chez l’animal que chez l’homme. De plus, on peut imaginer que les maladies rénales se développent de manière plus complexe chez l’humain, ou bien que la maladie initiale sous-jacente n’est jamais totalement guérie. D’autre part, chez l’homme, les maladies rénales se développent souvent tardivement, chez des personnes dont le rein a donc déjà « vécu » toute une vie de stress, d’exposition à des toxiques et à diverses maladies. Par comparaison, les modèles expérimentaux sont réalisés sur des animaux jeunes, élevés à l’abri de toute agression et soumis à un régime alimentaire contrôlé et dont le rein est sûrement plus apte à se régénérer. Enfin, un dernier paramètre à prendre en compte est la détection tardive des maladies rénales chez les patients. Ces maladies chroniques restent longtemps silencieuses et sont détectées à un stade où la fibrose est déjà installée depuis plusieurs années. Or la matrice accumulée est peu à peu modifiée, réticulée et devient au fil du temps plus résistante à la dégradation [39, 40]. La réversibilité nécessite donc, en plus de la diminution de la synthèse de MEC, une déstabilisation et une dégradation de la matrice déjà accumulée.
Un autre enjeu dans la recherche de nouveaux médicaments pour traiter la FTI est la notion de chronicité. En effet, ces maladies au long cours nécessitent des traitements qui dureront des dizaines d’années. Il est donc indispensable de mesurer le risque d’effets secondaires lorsque l’on bloque une voie pendant une aussi longue période de temps.
Comme nous l’avons discuté plus haut, de grands progrès ont été effectués ces dernières années afin d’élucider les mécanismes cellulaires et moléculaires mis en jeu dans ce processus de fibrose. Cela a permis d’identifier de nouvelles cibles thérapeutiques, intervenant à différentes étapes et impliquant les différents acteurs de la FTI, chacune présentant des avantages et des inconvénients (Tableau I). Ainsi certaines de ces stratégies, bien qu’elles aient montré des résultats encourageants chez l’animal (voir Encadré), n’ont toujours pas été évaluées chez l’homme. C’est le cas par exemple de l’administration d’HGF recombinant ou d’antagonistes du RB1 des kinines (Tableau I). D’autres, également prometteuses, présentent un risque trop important d’effets secondaires, comme par exemple le blocage du TGF-β ou du PDGF (Tableau I).
Étant donné la complexité du processus, il est clair que la molécule antifibrosante miracle n’existe pas. Aussi, en fonction du stade d’évolution auquel la fibrose aura été détectée, les futurs traitements devront-ils faire appel à la combinaison de plusieurs agents pharmacologiques agissant par des mécanismes moléculaires distincts et à des fenêtres thérapeutiques précises. ‡
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Remerciements
Julie Klein remercie la Fondation pour la recherche médicale et l’association 111 des Arts. Julie Klein, Jean-Loup Bascands et Joost P. Schanstra remercient le programme européen FP7 e-LICO pour leur soutien. Jean-Loup Bascands et Joost P. Schanstra sont soutenus par l’Inserm, la Direction régionale clinique (CHU de Toulouse) dans le cadre du programme d’interface et par l’ANR (ANR-07-PHYSIO-004-01).
Références
- Klahr S, Schreiner G, Ichikawa I. The progression of renal disease. N Engl J Med 1988 ; 318 : 1657-1666. [CrossRef] [PubMed] [Google Scholar]
- Nath KA. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am J Kidney Dis 1992 ; 20 : 1-17. [PubMed] [Google Scholar]
- Meguid El Nahas A, Bello AK. Chronic kidney disease: the global challenge. Lancet 2005 ; 365 : 331-340. [PubMed] [Google Scholar]
- Sean Eardley K, Cockwell P. Macrophages and progressive tubulointerstitial disease. Kidney Int 2005 ; 68 : 437-455. [CrossRef] [PubMed] [Google Scholar]
- Eddy AA. Progression in chronic kidney disease. Adv Chronic Kidney Dis 2005 ; 12 : 353-365. [CrossRef] [PubMed] [Google Scholar]
- Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest 2008 ; 118 : 3522-3530. [CrossRef] [PubMed] [Google Scholar]
- Wang Y, Wang YP, Zheng G, et al. Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int 2007 ; 72 : 290-299. [CrossRef] [PubMed] [Google Scholar]
- Velazquez P, Dustin ML, Nelson PJ. Renal dendritic cells: an update. Nephron Exp Nephrol 2009 ; 111 : e67-e71. [CrossRef] [PubMed] [Google Scholar]
- Heymann F, Meyer-Schwesinger C, Hamilton-Williams EE, et al. Kidney dendritic cell activation is required for progression of renal disease in a mouse model of glomerular injury. J Clin Invest 2009 ; 119 : 1286-1297. [CrossRef] [PubMed] [Google Scholar]
- Holdsworth SR, Summers SA. Role of mast cells in progressive renal diseases. J Am Soc Nephrol 2008 ; 19 : 2254-2261. [CrossRef] [PubMed] [Google Scholar]
- Grande MT, Lopez-Novoa JM. Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat Rev Nephrol 2009 ; 5 : 319-328. [CrossRef] [PubMed] [Google Scholar]
- Tesch GH. MCP-1/CCL2: a new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am J Physiol Renal Physiol 2008 ; 294 : F697-F701. [CrossRef] [PubMed] [Google Scholar]
- Giunti S, Tesch GH, Pinach S, et al. Monocyte chemoattractant protein-1 has prosclerotic effects both in a mouse model of experimental diabetes and in vitro in human mesangial cells. Diabetologia 2008 ; 51 : 198-207. [CrossRef] [PubMed] [Google Scholar]
- Eitner F, Bucher E, van Roeyen C, et al. PDGF-C is a proinflammatory cytokine that mediates renal interstitial fibrosis. J Am Soc Nephrol 2008 ; 19 : 281-289. [CrossRef] [PubMed] [Google Scholar]
- Strutz F, Zeisberg M, Ziyadeh FN, et al. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int 2002 ; 61 : 1714-1728. [CrossRef] [PubMed] [Google Scholar]
- Lautrette A, Li S, Alili R, et al. Angiotensin II and EGF receptor cross-talk in chronic kidney diseases: a new therapeutic approach. Nat Med 2005 ; 11 : 867-874. [CrossRef] [PubMed] [Google Scholar]
- Liu Y, Yang J. Hepatocyte growth factor: new arsenal in the fights against renal fibrosis? Kidney Int 2006 ; 70 : 238-240. [CrossRef] [PubMed] [Google Scholar]
- Liu Y. Hepatocyte growth factor in kidney fibrosis: therapeutic potential and mechanisms of action. Am J Physiol Renal Physiol 2004 ; 287 : F7-F16. [CrossRef] [PubMed] [Google Scholar]
- Schievenbusch S, Strack I, Scheffler M, et al. Profiling of anti-fibrotic signaling by hepatocyte growth factor in renal fibroblasts. Biochem Biophys Res Commun 2009 ; 385 : 55-61. [CrossRef] [PubMed] [Google Scholar]
- Giannopoulou M, Dai C, Tan X, et al. Hepatocyte growth factor exerts its anti-inflammatory action by disrupting nuclear factor-kappaB signaling. Am J Pathol 2008 ; 173 : 30-41. [CrossRef] [PubMed] [Google Scholar]
- Zeisberg M, Hanai J, Sugimoto H, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 2003 ; 9 : 964-968. [CrossRef] [PubMed] [Google Scholar]
- Zeisberg M, Kalluri R. Reversal of experimental renal fibrosis by BMP7 provides insights into novel therapeutic strategies for chronic kidney disease. Pediatr Nephrol 2008 ; 23 : 1395-1398. [CrossRef] [PubMed] [Google Scholar]
- Ruster C, Wolf G. Renin-angiotensin-aldosterone system and progression of renal disease. J Am Soc Nephrol 2006 ; 17 : 2985-2991. [CrossRef] [PubMed] [Google Scholar]
- Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol 2008 ; 214 : 199-210. [CrossRef] [PubMed] [Google Scholar]
- Mezzano SA, Ruiz-Ortega M, Egido J. Angiotensin II and renal fibrosis. Hypertension 2001 ; 38 : 635-638. [CrossRef] [PubMed] [Google Scholar]
- Carvajal G, Rodriguez-Vita J, Rodrigues-Diez R, et al. Angiotensin II activates the Smad pathway during epithelial mesenchymal transdifferentiation. Kidney Int 2008 ; 74 : 585-595. [CrossRef] [PubMed] [Google Scholar]
- Bascands JL, Schanstra JP, Couture R, et al. Les récepteurs de la bradykinine : de nouveaux rôles physiopathologiques. Med Sci (Paris) 2003 ; 19 : 1093-1100. [PubMed] [Google Scholar]
- Schanstra JP, Neau E, Drogoz P, et al. In vivo bradykinin B2 receptor activation reduces renal fibrosis. J Clin Invest 2002 ; 110 : 371-379. [PubMed] [Google Scholar]
- Klein J, Gonzalez J, Decramer S, et al. Blockade of the kinin B1 receptor ameloriates glomerulonephritis. J Am Soc Nephrol 2010 ; 21 : 1157-1164. [CrossRef] [PubMed] [Google Scholar]
- Klein J, Gonzalez J, Duchene J, et al. Delayed blockade of the kinin B1 receptor reduces renal inflammation and fibrosis in obstructive nephropathy. FASEB J 2009 ; 23 : 134-142. [CrossRef] [PubMed] [Google Scholar]
- Pradere JP, Gonzalez J, Klein J, et al. Lysophosphatidic acid and renal fibrosis. Biochim Biophys Acta 2008 ; 1781 : 582-587. [PubMed] [Google Scholar]
- Pradere JP, Klein J, Gres S, et al. LPA1 receptor activation promotes renal interstitial fibrosis. J Am Soc Nephrol 2007 ; 18 : 3110-3118. [CrossRef] [PubMed] [Google Scholar]
- Hirschberg R, Wang S. Proteinuria and growth factors in the development of tubulointerstitial injury and scarring in kidney disease. Curr Opin Nephrol Hypertens 2005 ; 14 : 43-52. [CrossRef] [PubMed] [Google Scholar]
- Strutz FM. EMT and proteinuria as progression factors. Kidney Int 2009 ; 75 : 475-481. [CrossRef] [PubMed] [Google Scholar]
- Fine LG, Norman JT. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: from hypothesis to novel therapeutics. Kidney Int 2008 ; 74 : 867-872. [CrossRef] [PubMed] [Google Scholar]
- Higgins DF, Kimura K, Iwano M, et al. Hypoxia-inducible factor signaling in the development of tissue fibrosis. Cell Cycle 2008 ; 7 : 1128-1132. [CrossRef] [PubMed] [Google Scholar]
- Fioretto P, Sutherland DE, Najafian B, et al. Remodeling of renal interstitial and tubular lesions in pancreas transplant recipients. Kidney Int 2006 ; 69 : 907-912. [CrossRef] [PubMed] [Google Scholar]
- Chatziantoniou C, Dussaule JC. Is kidney injury a reversible process? Curr Opin Nephrol Hypertens 2008 ; 17 : 76-81. [CrossRef] [PubMed] [Google Scholar]
- Fioretto P, Steffes MW, Sutherland DE, et al. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med 1998 ; 339 : 69-75. [CrossRef] [PubMed] [Google Scholar]
- Ronco P, Chatziantoniou C. Matrix metalloproteinases and matrix receptors in progression and reversal of kidney disease: therapeutic perspectives. Kidney Int 2008 ; 74 : 873-878. [CrossRef] [PubMed] [Google Scholar]
- Servais A, Meas-Yedid V, Morelon E, et al. Apports récents des techniques de quantification de la fibrose pour l’examen anatomo-pathologique en transplantation rénale. Med Sci (Paris) 2009 ; 25 : 945-950. [PubMed] [Google Scholar]
- Flamant M, Dussaule JC, Ardaillou R. Les effets profibrosants des peptides vasoactifs dans le rein et les vaisseaux passent-ils par la transactivation du facteur de croissance épidermique (EGF)? Med Sci (Paris) 2005 ; 21 : 461-463. [PubMed] [Google Scholar]
Liste des tableaux
Liste des figures
|
Figure 1 Les trois étapes de la FTI. Phase inflammatoire : en réponse à un stress, les cellules rénales résidentes sont activées et produisent des cytokines pro-inflammatoires et des chimiokines. Ceci induit le recrutement et l’infiltration de cellules inflammatoires qui produisent alors des radicaux libres oxygénés ou des cytokines pro-inflammatoires et profibrosantes. Phase d’apparition des myofibroblastes : le contexte inflammatoire mène à l’apparition des myofibroblastes par quatre voies principales : (a) à partir des cellules épithéliales tubulaires (TEM) ; (b) à partir des fibroblastes interstitiels ; (c) à partir des cellules endothéliales des capillaires (TEndM) et (d) à partir des péricytes. Phase d’accumulation de MEC : les myofibroblastes sont les cellules à l’origine de l’accumulation de MEC car ils synthétisent en excès les protéines matricielles mais ils sécrètent également les inhibiteurs des protéases dégradant la MEC. La présence de la FTI signe alors l’évolution vers la perte de la fonction rénale. |
| Dans le texte | |
|
Figure 2 Rôle des macrophages M1 et M2 dans la FTI. Le rôle duel des deux populations distinctes de macrophages est un phénomène reconnu dans le développement de la fibrose rénale. Ainsi, la stimulation chronique des macrophages de type M1 contribue à l’extension des lésions et au développement de la fibrose alors que les macrophages M2 permettent la diminution de l’inflammation, le remodelage matriciel et la régénération tissulaire. IFN : interféron ; IL- : interleukine ; TGF-β : transforming growth factor-β ; MEC : matrice extracellulaire ; TNF- α : tumor necrosis factor. |
| Dans le texte | |
|
Figure 3 Les mécanismes moléculaires de la progression de la fibrose rénale : un puzzle toujours plus complexe. Il s’agit ici d’avoir une vision un peu plus globale des principaux facteurs qui accélèrent (rouge) ou ou au contraire ralentissent (vert) la progression de la FTI. On voit ici à quel point la fibrose est un processus complexe et redondant. CTGF : connective tissue growth factor ; PDGF : platelet-derived growth factor ; FGF : fibroblast growth factor ; BMP-7 : bone morphogenetic protein -7 ; Ang II : angiotensine II ; LPA : acide lysophosphatidique ; RB1, RB2 : récepteurs des kinines ; HGF : hepatocyte growth factor ; EGF-R : récepteur de l’epidermal growth factor. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.