")
")
| Issue |
Med Sci (Paris)
Volume 22, Number 8-9, Août–Septembre 2006
|
|
|---|---|---|
| Page(s) | 733 - 738 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/20062289733 | |
| Published online | 15 août 2006 | |
Défaut d’exocytose des granules lytiques
Plusieurs causes, un même effet
Defect in lytic granule exocytosis: several causes, a same effect
Inserm U768, Hôpital Necker- Enfants-Malades, 149, rue de Sèvres, 75015 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Une réponse immune exagérée,ncontrôlée et le plus souvent fatale, connue sous le nom de syndrome hémophagocytaire (SH), est associée à un défaut de la fonction cytotoxique des lymphocytes T et natural killer (NK). Les anomalies moléculaires responsables, qui sont multiples, mettent en cause dans la plupart des cas un effecteur indispensable au fonctionnement de la machinerie lytique des lymphocytes. L’étude des lymphocytes cytotoxiques déficients en l’un ou l’autre de ces effecteurs apporte des éléments nouveaux quant à l’agencement des étapes clés de la sécrétion du contenu des granules lytiques au contact de la cellule cible. Des mécanismes moléculaires proches semblent contrôler la sécrétion vésiculaire au niveau des synapses immunologique et neurologique. D’autres effecteurs de la cytotoxicité ou du contrôle de l’homéostasie lymphocytaire à l’origine de SH doivent encore être caractérisés. Quant aux mécanismes précis de l’intervention de cette voie cytotoxique dans le maintien de l’homéostasie lymphocytaire (terminaison d’une réponse immune), ils demeurent à élucider.
Abstract
An in vivo disturbance of lymphocyte homeostasis occurs during the course of the hemophagocytic syndrome (HS). HS is a severe and often fatal syndrome resulting from potent and uncontrolled activation and proliferation of T-lymphocytes, mainly polyclonal CD8 lymphocytes, leading to excessive macrophage activation, high level of proinflammatory cytokine production and multiple deleterious effects. The onset of HS characterizes several inherited disorders in humans. In most of these conditions, the molecular defect impairs the granule-dependent cytotoxic activity of lymphocytes, thus highlighting the determinant role of this function in driving back the immune system to a state of equilibrium following infection. Several lines of evidence suggest that an increase in the expansion phase rather than a decrease in the contraction phase of the CD8+ T cells population characterizes the HS. Failure to kill antigen presenting cells through a transaction mechanism of cytotoxic cells should favor a sustained response, although the mechanism may be more complex than simple decrease of antigen load. Defect in the granule dependent cytotoxic function of lymphocytes result from perforin mutation in familial hemophagocytic lymphohistiocytosis type 2, from Munc13-4 (UNC13D) mutation in familial hemophagocytic lymphohistiocytosis type 3, from Rab27a mutation in Griscelli syndrome type 2, and from CHS/LYST mutation in Chediak- Higashi syndrome. The characterization of the molecular causes leading to these conditions identified Rab27a and Munc13-4 as two critical effectors of the exocytic machinery, required for the terminal transport/docking or priming of the cytotoxic granules, respectively. Different members of the Rab and Munc13 family of proteins are also used in neurotransmitter release at the neurological synapse, highlighting the similarity of the mechanisms regulating both secretory pathways. Future investigations regarding HS will continue to elucidate this exocytic pathway machinery and improve our understanding of how it finely regulates the immune response, an area that is likely to be useful for therapeutic intervention.
© 2006 médecine/sciences - Inserm / SRMS
La fonction cytotoxique des lymphocytes, un acteur de l’homéostasie lymphocytaire
Le système immunitaire est soumis principalement à deux types de sollicitations. Celle des antigènes du soi, d’une part, qui sont exprimés de façon permanente dans l’organisme, et pour lesquels le système immunitaire doit en permanence prévenir l’émergence de réponses auto-immunes. Un défaut de ce contrôle est à l’origine de maladies auto-immunes. Celle des antigènes « étrangers », d’autre part, notamment des pathogènes (virus, bactéries, parasites) qui vont activer, en règle de façon aiguë et transitoire, le système immunitaire. Un certain nombre de mécanismes sont mis en jeu à l’issue de la réaction immunitaire pour ramener à un état d’équilibre ce système immunitaire. Chez l’homme, un de ces mécanismes de contrôle dépend de la fonction cytotoxique des lymphocytes, dont le défaut conduit à un « syndrome hémophagocytaire » (SH) [1].
Au cours du SH, on observe une réponse immune exagérée et mal contrôlée, caractérisée par une prolifération et une activation très importante des lymphocytes T (surtout CD8) et des macrophages (Figure 1). Ces lymphocytes T activés sécrètent des quantités très importantes d’INFγ qui entraînent une activation macrophagique, comme en témoignent les images d’hémophagocytose caractéristiques (Figure 2) et la sécrétion importante de cytokines pro-inflammatoires (IFNγ, IL6, TNFα) [2]. Lymphocytes et macrophages activés infiltrent progressivement les différents organes, entraînant une nécrose tissulaire massive, une atteinte multiviscérale et le décès des patients en l’absence de traitement immunosuppresseur. Seule l’allogreffe de cellules souches hématopoïétiques permet de prévenir les récidives [3]. L’importance de la fonction cytotoxique des lymphocytes dans ce processus a été démontrée par l’existence d’une forme héréditaire de SH, la lymphohistiocytose familiale de type 2 (FHL2), qui résulte d’un défaut en perforine [4], un composant essentiel du granule lytique des cellules cytotoxiques qui induit, en synergie avec les granzymes A et B, la mort des cellules cibles par apoptose.
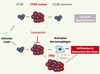 |
Figure 1. Modélisation de la réponse immune à un virus en situation normale et au cours du syndrome hémophagocytaire résultant d’un défaut d’activité cytotoxique des lymphocytes. En réponse à une infection virale, les lymphocytes spécifiques d’antigène subissent une expansion massive, et utilisent leur fonction cytotoxique et la sécrétion de cytokines telles que l’IFNγ pour éliminer efficacement les cellules infectées. Conjointement à la baisse de la quantité d’antigène disponible, la plupart des cellules T effectrices meurent, et il ne persiste que quelques cellules mémoires. Au contraire, un défaut d’activité cytotoxique dépendante des granules lytiques entraîne une expansion très importante de la population lymphocytaire effectrice, qui sécrète alors une très grande quantité d’INFγ. L’INFγ active secondairement les macrophages qui produisent des quantités importantes de cytokines pro-inflammatoires. Lymphocytes et macrophages activés envahissent différents organes, conduisant à une nécrose massive des tissus et des organes. |
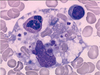 |
Figure 2. Image d’hémophagocytose. Macrophage activé ayant phagocyté in vivo des globules rouges, des plaquettes et un polynucléaire, processus dénommé « hémophagocytose ». |
Comment relier fonction cytotoxique et homéostasie lymphocytaire ?
L’étude chez la souris présentant un déficit en perforine permet d’approcher les mécanismes physiopathologiques à l’origine du SH. Les souris dont le gène de la perforine a été invalidé (modèle de lymphohistiocytose de type 2) ne développent pas spontanément de pathologie immunitaire, même lorsqu’elles sont maintenues en élevage non protégé. Cependant, lorsque ces souris sont infectées par certains virus comme le virus de la chorioméningite lymphocytaire (LCMV), elles développent tous les signes caractéristiques du syndrome hémophagocytaire, et meurent trois semaines après l’infection, avec une infiltration tissulaire massive des différents organes par des lymphocytes T et des macrophages activés [5, 6]. Dans les mêmes conditions, les souris sauvages contrôlent l’infection. Une étude fine des mécanismes en cause [6] met en évidence le rôle essentiel de la population lymphocytaire T CD8 et de la sécrétion très importante d’INFγ dans la genèse de ce processus. En effet, seule la déplétion en lymphocytes CD8 permet à ces souris déficientes en perforine de survivre après infection. La déplétion en lymphocytes T CD4 ou en cellules natural killer n’a pas d’effet. De même, parmi les différentes cytokines dont la sécrétion est augmentée au cours du syndrome hémophagocytaire (TNFα, INFγ, IL-6, INFα, IL-18), seule la neutralisation de l’INFγ permet une survie de ces souris.
Les mécanismes précis permettant à cette voie cytotoxique de contrôler la réponse immune restent cependant mal compris. Il est très probable qu’un des mécanismes utilisés est la lyse des cellules présentatrices d’antigène (APC) par les cellules T cytotoxiques (CTL) activées, son défaut favorisant la persistance de l’activation lymphocytaire. Plusieurs notions indiquent cependant que ce mécanisme n’est pas suffisant pour expliquer la dérégulation immunitaire très importante observée dans ce contexte, suggérant que d’autres mécanismes sont en jeu [6–8]. Un rôle de la voie cytotoxique dans l’apoptose des lymphocytes cytotoxiques n’a pas été pour l’instant clairement démontré. Une activité cytotoxique exercée en trans permettrait une fonction fratricide. Cette hypothèse s’appuie sur l’observation faite in vitro de la capacité des lymphocytes cytotoxiques à capturer des complexe MHC-peptide au moment du contact avec l’APC, les rendant transitoirement cibles pour d’autres CTL [9–11]. Il est également possible que cette fonction cytotoxique soit utilisée par des cellules T régulatrices, dont le défaut fonctionnel participerait à cette dérégulation homéostatique [12].
CHS, Rab27a et Munc13-4 désignent des étapes clés de l’exocytose des granules cytotoxiques
Le défaut de contrôle de l’homéostasie lymphocytaire que représente le SH est observé dans plusieurs pathologies héréditaires, en association ou non avec une hypopigmentation. C’est le cas en particulier du syndrome de Chediak-Higashi (CHS), du syndrome de Griscelli (GS) ou, encore, des différentes formes génétiques de lymphohistiocytose familiale (FHL). La caractérisation des causes moléculaires responsables de ces pathologies a permis d’identifier de nouveaux effecteurs impliqués dans le fonctionnement de la machinerie lytique des lymphocytes T et NK (Figure 3).
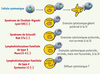 |
Figure 3. Distribution des granules cytotoxiques dans des CTL contrôles, et dans des CTL de patients présentants différents défauts génétiques liés à une anomalie de la fonction cytotoxique. A. Distribution des granules cytotoxiques (sphères vertes) sur le réseau de microtubules (lignes) dans des cellules T cytotoxiques (CTL) non conjuguées (à gauche). Après reconnaissance d’une cellule présentatrice d’antigène par une CTL et formation d’un conjugué entre les deux cellules (au milieu), les granules cytotoxiques se déplacent le long des microtubules vers le centre organisateur de microtubules (MTOC, en bleu), et polarisent avec lui vers le point de contact cellulaire ou se forme la synapse immunologique (SI). La sécrétion du contenu des granules induit l’apoptose de la cellule cible (à droite). B. Représentation des différents défauts cytotoxiques dans les CTL de patients présentant une mutation de Rab27a, de CHS/LYST, de Munc13-4 ou de la syntaxine 11, et conjugués avec une cellule cible. L’existence d’un défaut cytotoxique associé à une anomalie de la syntaxine 11 doit encore être démontrée. |
Un de ces effecteurs indispensables à la fonction cytotoxique est la protéine CHS, dont les anomalies provoquent le syndrome de Chediak-Higashi, ou son modèle murin, la souris beige. Un défaut en CHS est associé à la présence de granules cytotoxiques géants, capables d’être transportés vers la synapse immunologique (SI) au contact de la cellule cible, mais incapables de libérer leur contenu lytique [13]. Bien que l’identification de cette protéine remonte à une dizaine d’années, sa fonction précise reste inconnue. Sa très grande taille (> 400 kDa) explique en partie la difficulté de son étude. La présence de grosses granulations de nature lysosomale résultant d’un défaut en CHS suggère la participation de cette protéine dans les mécanismes de fusion ou de fission membranaire entre différents compartiments vésiculaires intracellulaires. Une expression de CHS est détectable dans de nombreux autres tissus : mélanocytes, fibroblastes, cellules tubulaires rénales, mais également cellules de Schwann et neurones. Son défaut rend compte de l’hypopigmentation qui caractérise ce syndrome, et peut-être également d’autres défauts fonctionnels, en particulier neurologiques, qui semblent apparaître au cours du temps [14].
Un deuxième effecteur clé de la machinerie lytique est la GTPase Rab27a, un membre de la famille des protéines Rab impliquée dans le transport vésiculaire. Son défaut est responsable du syndrome de Griscelli de type 2. Nous avons pu montrer que les lymphocytes cytotoxiques déficients en Rab27a étaient incapables de libérer le contenu de leurs granules lytiques, en raison, probablement, d’un défaut dans le transport terminal ou l’arrimage de ces granules à la membrane plasmique du lymphocyte, au niveau du contact cellulaire avec la cellule cible [15]. Comme celle de la protéine CHS, l’expression de Rab27a n’est pas restreinte aux lymphocytes. Au niveau des mélanocytes de la peau, Rab27a dirige le transport des mélanosomes contenant le pigment en périphérie de la cellule, et son déficit rend compte de l’hypopigmentation qui caractérise le syndrome de Griscelli [16]. Dans un certain nombre de tissus, comme dans le système nerveux, la fonction de Rab27a semble pouvoir être compensée par celle d’une isoforme très proche, Rab27b. Cela permet d’expliquer que l’expression de la pathologie soit essentiellement immunitaire et mélanocytaire.
De plus, l’observation de certains patients présentant l’anomalie pigmentaire caractéristique du syndrome de Griscelli, sans pour autant développer un syndrome hémophagocytaire, nous a permis de mettre en évidence la responsabilité d’effecteurs de la protéine Rab27a, dans ces formes phénotypiquement différentes du syndrome de Griscelli (types 1 et 3). Des anomalies de la mélanophiline [17], l’effecteur de Rab27a dans le mélanocyte, ou de son partenaire la myosine Va [18], observées chez les patients ayant un syndrome de Griscelli de type 3 et 1, respectivement, affectent l’interaction de ces protéines entre elles. Elles conduisent à un même défaut de transport des mélanosomes en périphérie des mélanocytes vers les kératinocytes adjacents, et donc à une hypopigmentation de la peau.
L’identification d’une des causes moléculaires à l’origine de la lymphohistiocytose familiale de type 3 (FHL3) nous a permis de caractériser un troisième effecteur de cette voie d’exocytose des granules lytiques, la protéine Munc13-4 [19]. La fonction de Munc13-4 est indispensable aux granules lytiques arrimés au niveau de la synapse immunologique pour leur permettre de fusionner avec la membrane plasmique, et ainsi libérer leur contenu lytique. Munc13-4 joue un rôle de facteur d’amorçage de la fusion membranaire, une fonction très proche de celle décrite au niveau de la synapse neurologique pour d’autres membres de la famille Munc13, qui semblent indispensables à l’exocytose des vésicules synaptiques contenant les neurotransmetteurs [20].
Récemment, une autre forme héréditaire du SH, la lymphohistiocytose familiale de type 4 (FHL4), a été associée à un défaut moléculaire de la syntaxine 11 [21]. Cette protéine appartient à la famille des SNARE (soluble N-ethyl-maleimide-sensitive factor attachment protein receptors), qui régulent la fusion membranaire entre les différents compartiments intracellulaires. Contrairement à tous les autres défauts moléculaires identifiés responsables d’un SH héréditaire, la syntaxine 11 pourrait ne pas être impliquée directement dans l’activité cytotoxique des lymphocytes T. Cette protéine semble d’ailleurs plus volontiers exprimée dans les cellules myéloïdes que lymphoïdes. Son expression dans les cellules présentatrices d’antigène pourrait lui permettre de participer indirectement à la fonction cytotoxique, par le biais du contact entre cellule présentatrice d’antigène et cellule T cytotoxique. Il est également possible que la syntaxine 11 régule l’homéostasie lymphocytaire par un mécanisme autre, révélé par cette pathologie immunitaire héréditaire, qu’il reste à caractériser.
Des mécanismes d’exocytose communs à la synapse immunologique et à la synapse neurologique
L’analogie entre les mécanismes qui régulent l’exocytose des vésicules à la synapse immunologique (SI) et ceux utilisés au niveau de la synapse neurologique (SN) mérite d’être soulignée (Figure 4A). Il faut remarquer que les contraintes fonctionnelles de ces deux types de synapses sont proches. Il s’agit dans les deux cas de rendre disponible rapidement, de façon transitoire, en un lieu précis et uniquement en ce lieu (au contact d’une « cible fonctionnelle »), le contenu actif de quelques vésicules parmi un large pool de vésicules en attente. Dans les deux situations, la sécrétion est régulée par un signal d’activation et doit pouvoir être répétée par une même cellule avec la même efficacité. Ce sont des protéines Rab, en particulier Rab3 au niveau de la SN et Rab27 au niveau de la SI qui, recrutées au niveau de la membrane vésiculaire, assurent le transport des vésicules à la synapse. À la SN, Rab3 interagit avec des partenaires spécifiques (comme RIM1, Rab3-interacting molecule 1) et des facteurs d’arrimage déjà présents au niveau de la zone active de la membrane présynaptique [22], assurant une localisation très précise des vésicules synaptiques au site de sécrétion. Les vésicules arrimées sont ensuite rendues compétentes pour la fusion au cours d’une étape d’amorçage dépendante d’un membre de la famille Munc13. Munc13-1 au niveau de la SN favorise l’ouverture conformationnelle de la syntaxine 1 et la formation en trans d’un complexe tripartite entre les protéines SNARE ancrées sur les vésicules (v-SNARE), et celles présentes au niveau de la membrane présynaptique (t-SNARE) [23, 24] (Figure 4A). La formation de ce complexe permet le rapprochement des deux membranes, puis la fusion, dépendante du calcium, des bicouches lipidiques. En agissant sur le quantum de vésicules prêtes à excréter leur contenu en réponse à l’influx calcique, Munc13-1 participe également à la plasticité neuronale [20].
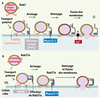 |
Figure 4. Modélisation du rôle des protéines Rab et Munc13 dans l’exocytose régulée au niveau de la synapse neurologique (SN) et de la synapse immunologique (SI). A. À la synapse neurologique, Rab3 est recrutée au niveau de la membrane de la vésicule présynaptique et participe à son transport jusqu’à la zone active de la synapse. Rab3 interagit alors avec son effecteur, RIM1 (Rab3-interacting molecule 1), prépositioné avec Munc13-1 au niveau de la membrane plasmique, permettant ainsi l’arrimage des vésicules à la membrane présynaptique. La fusion est déclenchée par l’action concertée de SNARE (soluble N-ethyl-maleimide-sensitive factor attachment protein receptors) et de Munc13-1. Les protéines SNARE, présentes sur chacun des partenaires de fusion, forment des ponts protéiques qui rapprochent les membranes en cours de fusion, une réaction déclenchée par Munc13-1. Après la fusion membranaire, les SNARE retrouvent leur état initial, avec l’aide d’autres effecteurs, pour constituer le cycle des SNARE. De même, Rab3 se détache de la membrane vésiculaire pour effectuer un nouveau cycle (cycle des Rab). B. Au niveau de la synapse immunologique, Rab27a semble recrutée au niveau de la vésicule cytotoxique et, en interagissant avec un (des) effecteur(s) non encore identifié(s), permettrait le transport et l’arrimage de vésicules à la SI. C’est par l’intermédiaire de ce transport vésiculaire que Munc13-4 atteint la SI nouvellement formée au point de contact entre CTL et cellule cible. Munc13-4 est impliqué dans l’étape d’amorçage de la fusion des vésicules, étape qui suit l’arrimage et précède la fusion à proprement parler des vésicules avec la membrane plasmique. Les membres de la famille SNARE impliqués à cette étape au niveau de la SI doivent encore être identifiés. |
L’étude du mécanisme conduisant à l’exocytose des granules cytotoxiques au niveau de la SI laisse entrevoir une séquence d’événements similaire : la stimulation des lymphocytes cytotoxiques, par reconnaissance de l’antigène que leur présente la cellule cible, induit la polarisation du centre organisateur des microtubules (MTOC), de l’appareil de Golgi et des granules cytotoxiques vers la zone de contact membranaire entre cellule cytotoxique et cellule présentatrice d’antigène [10]. C’est au cours de cette activation que Rab27a serait recrutée sur les granules cytotoxiques polarisés (Figure 4B). Le transport et l’arrimage des granules cytotoxiques à la SI fait intervenir un effecteur de Rab27a encore inconnu. De même, par analogie avec les mécanismes décrits au niveau de la SN, Munc13-4 devrait agir au niveau de la SI, en aval de Rab27a, pour préparer la phase de fusion membranaire du granule avec la membrane plasmique, en interagissant avec un membre de la famille des SNARE. L’interaction entre Munc13-4 et Rab27a récemment mise en évidence [25] pourrait également être impliquée dans cette étape. Enfin, une caractéristique essentielle de la SI est sa nature transitoire, et surtout sa localisation éminemment labile qui dépend du lieu de contact avec la cellule cible, une différence notable avec la SN. C’est peut-être pour s’adapter à cette variabilité que Munc13-4 n’est pas prélocalisée au niveau de la membrane plasmique, comme l’est Munc13-1, mais est transportée par le biais du trafic vésiculaire vers le site de sécrétion formé de novo.
Conclusions et perspectives
La sécrétion des granules lytiques des lymphocytes est une fonction essentielle du système immunitaire, en raison de sa participation à la défense de l’organisme contre des cellules tumorales ou infectées, mais également dans la régulation de la réponse immune. D’autres effecteurs de cette voie cytotoxique devraient être à l’origine de SH non encore caractérisés sur le plan moléculaire. Inversement, la caractérisation moléculaire de SH non associés à un défaut cytotoxique devrait révéler d’autre(s) voie(s) de contrôle de l’homéostasie lymphocytaire. Les mécanismes régulateurs précis mis en œuvre restent imparfaitement compris. Cependant, l’importance de la sécrétion exagérée d’IFNγ dans le processus du SH incite à tenter de cibler les effecteurs de cette molécule dans le traitement des SH. Enfin, la compréhension des mécanismes physiopathologiques à l’origine des SH héréditaires devrait aider à mieux appréhender les mécanismes pouvant conduirent au développement d’un syndrome hémophagocytaire acquis, tel qu’il peut être observé au cours de certaines infections virales comme l’infection par le virus Epstein-Barr ou, comme cela a très récemment été proposé, par le virus influenza responsable de la grippe aviaire [26, 27].
Article reçu le 24 avril 2006, accepté le 16 juin 2006.
Remerciements
Nous tenons à remercier l’Inserm, l’ANR et l’AP-HP, qui soutiennent nos travaux.
Références
- Menasche G, Feldmann J, Fischer A, de Saint Basile G. Primary hemophagocytic syndromes point to a direct link between lymphocyte cytotoxicity and homeostasis. Immunol Rev 2005; 203 : 165–79. [Google Scholar]
- Henter JI, Elinder G, Soder O, et al. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood 1991; 78 : 2918–22. [Google Scholar]
- Ouachee-Chardin M, Elie C, de Saint Basile G, et al. Hematopoietic stem cell transplantation in hemophagocytic lymphohistiocytosis : a single-center report of 48 patients. Pediatrics 2006; 117 : e743–50. [Google Scholar]
- Stepp S, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 1999; 286 : 1957–9. [Google Scholar]
- Matloubian M, Suresh M, Glass A, et al. A role for perforin in downregulating T-cell responses during chronic viral infection. J Virol 1999; 73 : 2527–36. [Google Scholar]
- Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH) : CD8+ T cells and interferon gamma are essential for the disorder. Blood 2004; 104 : 735–43. [Google Scholar]
- Badovinac VP, Harty JT. CD8(+) T-cell homeostasis after infection : setting the « curve ». Microbes Infect 2002; 4 : 441–7. [Google Scholar]
- Kagi D, Odermatt B, Mak TW. Homeostatic regulation of CD8+ T cells by perforin. Eur J Immunol 1999; 29 : 3262–72. [Google Scholar]
- Huang JF, Yang Y, Sepulveda H, et al. TCR-mediated internalization of peptide-MHC complexes acquired by T cells. Science 1999; 286 : 952–4. [Google Scholar]
- Stinchcombe JC, Bossi G, Booth S, Griffiths GM. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity 2001; 15 : 751–61. [Google Scholar]
- Hudrisier D, Riond J, Mazarguil H, et al. Cutting edge : CTLs rapidly capture membrane fragments from target cells in a TCR signaling-dependent manner. J Immunol 2001; 166 : 3645–9. [Google Scholar]
- Grossman WJ, Verbsky JW, Barchet W, et al. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 2004; 21 : 589–601. [Google Scholar]
- Nagle DL, Karim MA, Woolf EA, et al. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet 1996; 14 : 307–11. [Google Scholar]
- Tardieu M, Lacroix C, Neven B, et al. Progressive neurologic dysfunctions 20 years after allogeneic bone marrow transplantation for Chediak-Higashi syndrome. Blood 2005; 106 : 40–2. [Google Scholar]
- Menasche G, Pastural E, Feldmann J, et al. Mutations in Rab27a cause Griscelli syndrome associated with hemophagocytic syndrome. Nat Genet 2000; 25 : 173–6. [Google Scholar]
- Bahadoran P, Aberdam E, Mantoux F, et al. Rab27a. A key to melanosome transport in human melanocytes. J Cell Biol 2001; 152 : 843–50. [Google Scholar]
- Menasche G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest 2003; 112 : 450–6. [Google Scholar]
- Pastural E, Barrat FJ, Dufourcq-Lagelouse R, et al. Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin-Va gene. Nat Genet 1997; 16 : 289–92. [Google Scholar]
- Feldmann J, Callebaut I, Raposo G, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 2003; 115 : 461–73. [Google Scholar]
- Rosenmund C, Sigler A, Augustin I, et al. Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron 2002; 33 : 411–24. [Google Scholar]
- zur Stadt U, Schmidt S, Kasper B, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet 2005; 14 : 827–34. [Google Scholar]
- Betz A, Thakur P, Junge HJ, et al. Functional interaction of the active zone proteins Munc13-1 and RIM1 in synaptic vesicle priming. Neuron 2001; 30 : 183–96. [Google Scholar]
- Chen YA, Scheller RH. SNARE-mediated membrane fusion. Nat Rev Mol Cell Biol 2001; 2 : 98–106. [Google Scholar]
- Richmond JE, Weimer RM, Jorgensen EM. An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature 2001; 412 : 338–41. [Google Scholar]
- hirakawa R, Higashi T, Kondo H, et al. Purification and functional analysis of a Rab27 effector munc 13-4 using a semi-intact platelet dense-granule secretion assay. Meth Enzymol 2005; 403 : 778–88. [Google Scholar]
- enter JI, Chow CB, Leung CW, Lau YL. Cytotoxic therapy for severe avian influenza A (H5N1) infection. Lancet 2006; 367 : 870–3. [Google Scholar]
- sieh SM, Chang SC. Cutting edge : insufficient perforin expression in CD8+ T cells in response to hemagglutinin from avian influenza (H5N1) virus. J Immunol 2006; 176 : 4530–3. [Google Scholar]
Liste des figures
|
Figure 1. Modélisation de la réponse immune à un virus en situation normale et au cours du syndrome hémophagocytaire résultant d’un défaut d’activité cytotoxique des lymphocytes. En réponse à une infection virale, les lymphocytes spécifiques d’antigène subissent une expansion massive, et utilisent leur fonction cytotoxique et la sécrétion de cytokines telles que l’IFNγ pour éliminer efficacement les cellules infectées. Conjointement à la baisse de la quantité d’antigène disponible, la plupart des cellules T effectrices meurent, et il ne persiste que quelques cellules mémoires. Au contraire, un défaut d’activité cytotoxique dépendante des granules lytiques entraîne une expansion très importante de la population lymphocytaire effectrice, qui sécrète alors une très grande quantité d’INFγ. L’INFγ active secondairement les macrophages qui produisent des quantités importantes de cytokines pro-inflammatoires. Lymphocytes et macrophages activés envahissent différents organes, conduisant à une nécrose massive des tissus et des organes. |
| Dans le texte | |
|
Figure 2. Image d’hémophagocytose. Macrophage activé ayant phagocyté in vivo des globules rouges, des plaquettes et un polynucléaire, processus dénommé « hémophagocytose ». |
| Dans le texte | |
|
Figure 3. Distribution des granules cytotoxiques dans des CTL contrôles, et dans des CTL de patients présentants différents défauts génétiques liés à une anomalie de la fonction cytotoxique. A. Distribution des granules cytotoxiques (sphères vertes) sur le réseau de microtubules (lignes) dans des cellules T cytotoxiques (CTL) non conjuguées (à gauche). Après reconnaissance d’une cellule présentatrice d’antigène par une CTL et formation d’un conjugué entre les deux cellules (au milieu), les granules cytotoxiques se déplacent le long des microtubules vers le centre organisateur de microtubules (MTOC, en bleu), et polarisent avec lui vers le point de contact cellulaire ou se forme la synapse immunologique (SI). La sécrétion du contenu des granules induit l’apoptose de la cellule cible (à droite). B. Représentation des différents défauts cytotoxiques dans les CTL de patients présentant une mutation de Rab27a, de CHS/LYST, de Munc13-4 ou de la syntaxine 11, et conjugués avec une cellule cible. L’existence d’un défaut cytotoxique associé à une anomalie de la syntaxine 11 doit encore être démontrée. |
| Dans le texte | |
|
Figure 4. Modélisation du rôle des protéines Rab et Munc13 dans l’exocytose régulée au niveau de la synapse neurologique (SN) et de la synapse immunologique (SI). A. À la synapse neurologique, Rab3 est recrutée au niveau de la membrane de la vésicule présynaptique et participe à son transport jusqu’à la zone active de la synapse. Rab3 interagit alors avec son effecteur, RIM1 (Rab3-interacting molecule 1), prépositioné avec Munc13-1 au niveau de la membrane plasmique, permettant ainsi l’arrimage des vésicules à la membrane présynaptique. La fusion est déclenchée par l’action concertée de SNARE (soluble N-ethyl-maleimide-sensitive factor attachment protein receptors) et de Munc13-1. Les protéines SNARE, présentes sur chacun des partenaires de fusion, forment des ponts protéiques qui rapprochent les membranes en cours de fusion, une réaction déclenchée par Munc13-1. Après la fusion membranaire, les SNARE retrouvent leur état initial, avec l’aide d’autres effecteurs, pour constituer le cycle des SNARE. De même, Rab3 se détache de la membrane vésiculaire pour effectuer un nouveau cycle (cycle des Rab). B. Au niveau de la synapse immunologique, Rab27a semble recrutée au niveau de la vésicule cytotoxique et, en interagissant avec un (des) effecteur(s) non encore identifié(s), permettrait le transport et l’arrimage de vésicules à la SI. C’est par l’intermédiaire de ce transport vésiculaire que Munc13-4 atteint la SI nouvellement formée au point de contact entre CTL et cellule cible. Munc13-4 est impliqué dans l’étape d’amorçage de la fusion des vésicules, étape qui suit l’arrimage et précède la fusion à proprement parler des vésicules avec la membrane plasmique. Les membres de la famille SNARE impliqués à cette étape au niveau de la SI doivent encore être identifiés. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.