")
")
| Issue |
Med Sci (Paris)
Volume 19, Number 2, Février 2003
|
|
|---|---|---|
| Page(s) | 173 - 186 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2003192173 | |
| Published online | 15 February 2003 | |
Cycle cellulaire et points de contrôle en oncologie : nouvelles cibles thérapeutiques
New anticancer therapeutic targets in the cell cycle and cell cycle checkpoints
Laboratoire de pharmacologie moléculaire, Center for Cancer Research, National Cancer Institute, Bldg 37, Room 5068, NIH, Bethesda, MD 20892-4255, États-Unis
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
La découverte de l’existence des gènes Cdc (cell division cycle) et des complexes cycline:Cdk, et de leur conservation dans l’évolution a valu à Leland H. Hartwell, Paul M. Nurse et R. Timothy Hunt le prix Nobel de physiologie et de médecine en 2001. Cet article fait le point des connaissances actuelles sur les molécules de régulation du cycle cellulaire, et met en avant l’importance de ces connaissances dans la compréhension des mécanismes responsables des cancers et dans le développement de nouvelles thérapeutiques en oncologie.
Abstract
Leland H. Hartwell, Paul M. Nurse et R. Timothy Hunt just received the Nobel price for their discovery of the molecular components of the cell cycle and cell cycle checkpoints. This review is an update of the molecular networks driving the cell cycle and its regulation, and of the importance of this knowledge for understanding the mechanisms driving oncogenesis and therapeutic developments.
© 2003 médecine/sciences - Inserm / SRMS
Les composantes moléculaires du cycle cellulaire et de ses points de surveillance (checkpoints) sont importantes en cancérologie. En effet, leur connaissance peut servir à détecter les prédispositions familiales, à évaluer individuellement les tumeurs et à identifier leurs déterminants moléculaires, ainsi qu’à mettre au point de nouvelles approches thérapeutiques, éventuellement adaptées à chaque type de tumeurs en fonction de ses caractéristiques moléculaires.
Cet article propose un panorama des voies moléculaires régulatrices du cycle cellulaire et de ses points de surveillance, une discussion sur les bases rationnelles de l’utilisation d’inhibiteurs et une description des agents thérapeutiques dirigés contre différents sites d’action. Pour des raisons éditoriales, seules sont référencées les revues préexistantes et les publications les plus récentes, en particulier pour celles qui concernent les inhibiteurs des points de surveillance du cycle cellulaire [1–5].
En raison de la richesse des interactions moléculaires, une convention rigoureuse et concise a été développée, qui est résumée dans la Figure 1 (pour plus de détails, voir [6] et http://discover.nci.nih.gov/kohnk/interaction_maps.html). Les lecteurs sont invités à prendre le temps d’explorer cette approche, même si celle-ci peut paraître relativement abstraite au premier abord. La Figure 3 (transition G1→S) et la Figure 4 (transition G2→M) présentent des coordonnées cartographiques introduites pour faciliter la localisation des molécules mentionnées dans le texte (par exemple, la protéine pRb est en position B.4 dans la Figure 3).
 |
Figure 1. Symboles et conventions utilisés [6]. (a) La double flèche indique que les protéines A et B se lient l’une à l’autre. Le point sur la ligne représente le complexe A:B. (b) Représentation de complexes multimoléculaires: x est [A:B] et y est [(A:B):C]. Cette notation peut être étendue à un nombre plus grand de molécules dans un complexe. (c) Modification covalente de la protéine A. La flèche indique que A peut exister à l’état phosphorylé. Le point représente la protéine A phosphorylée. (d) Clivage d’une liaison covalente: déphosphorylation de A par une phosphatase. (e) Conversion stoechiométrique de A à B. Dégradation de A (c’est-à-dire conversion de A en son produit de dégradation Ø). (f) Stimulation enzymatique d’une réaction. (g) Symbole général de stimulation. (h) Symbole général d’inhibition. (i) Symbole d’activation de la transcription. |
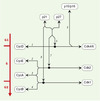 |
Figure 2. Associations moléculaires entre cyclines, Cdk et CKI. Il existe au moins neuf Cdk (cyclin-dependent kinases), les protéines Cdk 1 à 7 ayant été clairement impliquées dans le cycle cellulaire. Cdk7, Cdk8 et Cdk9 sont impliquées dans la transcription. Il existe au moins 15 cyclines (cyclines A à T) dont l’expression varie durant le cycle cellulaire. Les protéines Cdk sont activées par leur liaison aux cyclines et inactivées par leur liaison aux CKI. L’échelle à gauche représente les phases du cycle cellulaire durant lesquelles les complexes cyclines:Cdk sont actifs. (1) La cycline D (CycD) se lie à Cdk4 ou Cdk6 (le complexe bimoléculaire est représenté par le point noir, voir Figure 1). (2) La cycline E (CycE) se lie à Cdk2. (3) La cycline A (CycA) se lie à Cdk2 durant la phase S et à Cdk1 (appelée aussi Cdc2) en fin de phase S et durant la phase G2. (4) La cycline B (CycB) se lie à Cdk1. (5) Les CKI (cyclin-dependent kinase inhibitors) p16INK4a ou p15INK4b entrent en compétition avec la cycline D pour se fixer à Cdk4 ou Cdk6, ce qui empêche l’activation de ces kinases. (6) p21Waf1/Cip1 et p27kip1 se lient aux complexes cycline D:Cdk4 ou cycline D:Cdk6. Lorsque p21cip1 et p27kip1 sont en excès, il en résulte une inhibition. En revanche, de faibles concentrations de p21Waf1/Cip1 pourraient avoir un effet stimulant en empêchant la dégradation de la cycline D. (7) p27kip1 ou p21Waf1/Cip11 se lient aux complexes cycline E:Cdk2 ou cycline A:Cdk2 et inhibent leur activité kinase. (8) p21Waf1/Cip1 peut également se lier au complexe cycline B:Cdk1 et inhiber son activité. |
 |
Figure 3. Interactions moléculaires de la transition G1→S et du contrôle du facteur de transcription E2F. Les symboles sont définis par Kohn [6]. (1) [coordonnées D3] La transition G1→S implique les gènes contrôlés par E2F et la cycline E. (2) [C5] Les gènes dépendants de E2F codent pour des protéines régulatrices telles que la cycline E et la protéine Myc, et des gènes dont les produits sont des enzymes impliquées dans la réplication (non incluses sur le schéma). Ces gènes sont activés par les complexes E2F1:DP1 (et d’autres hétérodimères E2F) et négativement réglés lorsque E2F est lié à pRb. Cette dernière possède donc un rôle majeur. (3) [B4] La liaison de pRb à l’hétérodimère d’E2F est inhibée lorsque pRb est phosphorylée. pRb est également inhibée par les protéines virales E1A et l’antigène grand T (SV40-T). (4) [B5] La dégradation de pRb est stimulée par la protéine E7 du virus du papillome. (5) [B4] pRb est phosphorylée par les complexes cycline D:Cdk4/6 et cycline E:Cdk2. Ces deux complexes kinases sont nécessaires pour phosphoryler pRb complètement et bloquer l’interaction entre pRb et E2F. (6)[B.2] Cdk4 et Cdk6 sont actives quand elles sont couplées à la cycline D. (7)[B2] Cdk4 et Cdk6 peuvent être phosphorylées à des sites qui stimulent ou bloquent leur activité. (8)[B2] Les CKI p15 ou p16 peuvent se fixer à Cdk4 ou Cdk6 et bloquer leur fixation de la cycline D. Cela a pour effet d’inactiver les kinases. (9)[A2] Le TGFβ active la transcription de p15 par la voie des Smad (non montré). (10)[B2] La protéine Cdk6 peut être inactivée par déplacement de la cycline D par la protéine virale KSHV/HHV8 du virus du sarcome de Kaposi. (11)[B2] Les hétérodimères cycline D:Cdk4/6 peuvent se lier à p21cip1 ou p27kip1, ce qui a pour effet de les stabiliser. (12)[B2] La cycline D a une demi-vie courte, d’environ 30 minutes. Elle est dégradée dans le protéasome après ubiquitinylation. (13)[B.2] La protéine p53 active p21cip1. (14)[B2] p21cip1 se fixe à PCNA, un co-facteur de l’ADN polymérase. (15)[C.2] p21cip1 ou p27kip1 se lient aux complexes cycline E:Cdk2 et bloquent leur activité kinase (par opposition aux complexes cycline D:Cdk4/6 (voir [11] ci-dessus). (16)[C2] p27kip1 est phosphorylée par la cycline E:Cdk2. p27kip1 est donc à la fois un substrat et un inhibiteur des complexes kinases cycline E. (17)[C1] p27kip1 est dégradée après phosphorylation. (18)[C2] La liaison de la protéine virale KSHV/HHV8 stimule la dégradation de p27kip1. (19)[C2-3] Les complexes cycline E:Cdk2 (et cycline D:Cdk4/6) sont phosphorylés au niveau de sites d’activation et d’inhibition. (20)[A3] Cdk2, Cdk4 et Cdk6 sont phosphorylées et activées par la cycline H:Cdk7 (encore connue sous le nom de CAK), qui est un composant du facteur de transcription TFIIH. (21)[B3] Cdk2, Cdk4 et Cdk6 sont activées par déphosphorylation par la phosphatase Cdc25A. (22)[A4] Cdc25A est activée par phosphorylation par Raf1 dans la voie de signalisation Ras. (23)[A5] La transcription du gène de Cdc25A est activée par les complexes c-Myc:Max. (24)[D2-3] Le complexe cycline A:Cdk2 phosphoryle E2F1 et DP1, ce qui produit la dissociation du complexe E2F1:DP1, avec pour effet d’arrêter l’activité de E2F en fin de phase S. Les conventions utilisées pour le choix des couleurs sont les suivantes; en rouge, modifications covalentes et voies de transcription; en vert, modifications enzymatiques; en bleu: effets stimulants ou inhibiteurs; en noir, liaisons intermoléculaires; lettres rouges: inputs et outputs. |
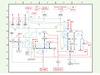 |
Figure 4. Interactions moléculaires de la transition G2→M. (1)[coordonnées E3] La transition G2→M, qui prépare à la mitose, est principalement contrôlée par le complexe kinase cycline B:Cdk1. (2)[D2] L’activité kinase de Cdk1 est contrôlée par des phosphorylations spécifiques. La phosphorylation de la thréonine 161 est nécessaire pour son activité, alors que la phosphorylation de la thréonine 14 et de la tyrosine 15 sont inhibitrices. (3)[D2] La thréonine 161 de Cdk1 est phosphorylée par la cycline H:Cdk7 (CAK) (voir Figure 3, phosphorylations stimulantes de Cdk2 et Cdk4/6). (4)[D2] La tyrosine kinase nucléaire Wee1 phosphoryle spécifiquement la tyrosine 15 de Cdk1 (et de Cdk2) pendant les phases S et G2, alors que la kinase cytoplasmique Myt1 a une double spécificité pour la thréonine 14 et la tyrosine15 de Cdk1. (5)[C2] Inversement, ces résidus sont déphosphorylés par la phosphatase Cdc25C. (6)[C1] Cdc25C peut être activée par phosphorylation par la Polo kinase Plk1 et par le complexe cycline B:Cdk1, ce qui produit une boucle de contrôle positif pour l’activation de ce complexe. (7)[C1] La phosphorylation de Cdc25C sur la sérine 216 stimule sa liaison à la protéine 14–3–3σ, avec pour résultat une inhibition de sa phosphatase. La liaison de 14–3–3σ à la kinase Weel stimule également l’activité de Wee1 (non montré) [45]. La protéine 14–3–3σ a donc au moins deux mécanismes régulateurs négatifs sur la progression G2→M. (8)[C1-2] La phosphorylation de Cdc25C sur la sérine 216 peut être catalysée par Chk1, Chk2 ou Plk3 (voir annotation 25 ci-dessous). (9)[B1] Chk1 et Chk2 sont activées par phosphorylation. (10)[B2-3] Chk1 semble être principalement phosphorylée par ATR, alors que Chk2 peut être phosphorylée par ATM (ou ATR). (11)[A3] ATR est activée lors de la détection de blocages des fourches de réplication de l’ADN produites par les ultraviolets (UV) ou la camptothécine (CPT). (12)[A4] ATM est activée par des coupures double-brin de l’ADN (DSB) produites par divers agents tels que les radiations ionisantes (IR) ou l’inhibiteur de topo-isomérase II, l’étoposide (VP16). (13)[A-B2-3] La caféine inhibe ATM et ATR. (14)[A2] L’UCN-01 inhibe une variété de protéine kinases, dont Chk1 et Chk2, mais aussi Wee1. Cet effet est indirect. (15)[B4] ATM peut phosphoryler p53 sur sa sérine 15, ce qui stabilise p53 en réduisant sa dégradation. (16)[B3-4] Chk1 et Chk2 phosphorylent p53 sur la sérine 20, ce qui a pour effet de bloquer la liaison de Mdm2. (17)[C5] Mdm2 est un antagoniste de p53, agissant d’une part en l’ubiquitinylant, ce qui déclenche sa dégradation, et d’autre part en bloquant son activité transcriptionnelle. (18)[D5] Mdm2 se fixe à p14Arf, ce qui a pour effet de diminuer la dégradation de p53 liée à la fixation de Mdm2. (19)[E4] Mdm2 se fixe à E2F1 et stimule sa dégradation. Cela bloquerait l’activation des gènes spécifiques de la phase S (voir Figure 3). (20)[D5] La protéine p53 active de nombreux gènes, y compris ceux de Mdm2, p21cip1, Gadd45, 14–3–3 et Bax. (21)[C5] La p53 inhibe d’autre gènes, comme ceux de la cycline B1 et de Cdk1. (22)[C2-3] p21cip1 inhibe plusieurs complexes cycline-kinase (voir Figure 2), comme la cycline H:Cdk7 (CAK). (23)[C-D3] Gadd45 et 14–3–3σ se fixent au complexe cycline B:Cdk1 et bloquent son activité kinase. (24)[A2] Chk1 est activée par les UV et les anomalies de réplication, alors que Chk2 est activée par les radiations ionisantes (IR). (25)[B1] Plk3 est activée par ATM et ATR et, comme Chk1 et Chk2, phosphoryle p53 sur la sérine 20 et Cdc25C sur la sérine 216. Plk3 peut être activée par la doxorubicine, les UV, et le stress oxydatif (H2O2). (26)[C4] Un sillon dans la région N-terminale de Mdm2 lie un segment peptidique dans la région N-terminale de p53, ce qui stimule la dégradation de p53 après ubiquitinylation (26a)[C4]. (27)[D4] ATM inactive l’interaction entre Mdm2 et p53 par phosphorylation de Mdm2. |
Régulation moléculaire du cycle cellulaire
Un des mécanismes fondamentaux de la cancérogenèse est la prolifération incontrôlée des cellules tumorales. La majorité des cellules d’un organisme normal ne se divisent pas: elles sont dites quiescentes (phase G0 pour Gap 0). Le passage des cellules dans une phase de prolifération nécessite le déclenchement du cycle cellulaire, défini par Arthur Pardee, il y a environ 35 ans, comme le point de restriction (restriction point) ou point de non-retour. Passé celui-ci, la cellule progresse dans le cycle cellulaire, qui se décompose en quatre phases: G1 (Gap 1 ou pause 1, qui précède la synthèse d’ADN), S (synthèse de l’ADN), G2 (Gap 2 ou pause 2) qui précède la phase M (mitose).
Les protéine kinases dépendantes des cyclines (Cdk)
La prolifération cellulaire nécessite l’action coordonnée de complexes protéiques constitués de cyclines et de leurs partenaires, les Cdk (cyclin-dependent protein kinases) (Figure 2), ces dernières n’étant actives que lorsqu’elles ont fixé leur partenaire cycline. Au niveau moléculaire, le passage du point de restriction nécessite l’activation d’au moins deux types de complexes cycline-kinases, la cycline D:Cdk4-6 et la cycline E/A:Cdk2 (Figures 2 et 3), qui engagent les cellules à transiter de la phase G1 à la phase S et à dupliquer leurs centrosomes. Pendant toute la phase de synthèse de l’ADN, la Cdk2 est active sous forme de complexes avec la cycline E ou la cycline A. La transition G2→M est quant à elle sous le contrôle du complexe cycline B:Cdk1.
Un des éléments régulateurs du cycle cellulaire est donc la variation cyclique et coordonnée de l’activité des différents complexes cycline: Cdk. Ces fluctuations sont dues à une régulation des Cdk par phosphorylation et à un contrôle des niveaux de cyclines dans le noyau cellulaire. Les signaux mitogènes (par exemple sous le contrôle des gènes Myc et Ras) induisent la synthèse de cycline D. Les gènes codant pour les cyclines E et A sont quant à eux sous le contrôle positif des complexes transcriptionnels E2F (Figure 3, C4). Inversement, c’est par leur dégradation que les niveaux de cyclines sont contrôlés négativement à des points précis du cycle cellulaire. Par exemple, la transition G2→M nécessite la dégradation de la cycline B par le complexe MPF (mitosis promoting factor). Par ailleurs, dans des conditions normales, la dégradation de la cycline E par ubiquitinylation et protéolyse dans le protéasome joue un rôle important dans la progression de la prolifération cellulaire [7]. Amplification génétique ou déficience des voies de dégradation pourraient être toutes deux à l’origine d’une élévation des niveaux de cyclines D et E en particulier, et donc d’une activation des complexes cyclines:Cdk dans certains cancers. Une des molécules récemment impliquées est l’ubiquitine ligase hCdc4 qui pourrait agir comme gène suppresseur de tumeurs [7] par dégradation des cyclines.
La distribution cellulaire des cyclines est également un élément régulateur de l’activité des complexes cycline:Cdk. Par exemple, pendant l’interphase, la cycline B1 est principalement dans le cytoplasme. En revanche, au moment de la transition G2→M, la phosphorylation de la cycline B1 bloque sa sortie du noyau, ce qui concentre les complexes cycline B1:Cdk1 au niveau nucléaire et leur permet d’activer leurs substrats.
L’activité kinase des Cdk est modulée par phosphorylation. Les Cdk sont activées par la phosphorylation d’une thréonine dans la portion carboxy-terminale des Cdk par les kinases CAK (pour cyclin activated kinase, ou complexe CycH:Cdk7) (Figure 4, D-E2) et c-TAK1. Le résidu cible est la thréonine 161 pour Cdk1, 160 pour Cdk2 et 172 pour Cdk4. Inversement, les Cdk sont inactivées par phosphorylation du site ATP dans leur portion aminoterminale (tyrosine 15 et thréonine 14) par les kinases Wee1 et Myt1 (Figure 3, C3; Figure 4, C-D2). L’activation de la prolifération cellulaire par la kinase Akt (ou protéine kinase B, PKB) a très récemment été attribuée à une phosphorylation qui inactive Myt1, ce qui empêche l’inactivation de la Cdk1.
L’activation des Cdk peut également être obtenue par déphosphorylation de leur résidu tyrosine 15 ou thréonine 14 par les phosphatases de la famille Cdc25. Les trois phosphatases de la famille Cdc25 (Cdc25A, Cdc25B et Cdc25C) sont très similaires (l’homologie est de l’ordre de 50 % au niveau des séquences de protéines) [8], bien qu’elles fonctionnent à différents points du cycle cellulaire. La protéine Cdc25A est nécessaire pour l’entrée en phase S (Figure 3, A4), alors que Cdc25B et Cdc25C assurent le passage en mitose (Figure 3, A4; Figure 4, C1-2). Cdc25C peut être activée par phosphorylation par la Polo kinase Plk1 et le complexe cycline B1:Cdk1 (Figure 4, C1). Ainsi, il a été proposé que Cdk1 serait soumise à une boucle de rétro-activation ayant pour finalité de produire un passage sans retour de la phase G2 à la mitose. Au stade de la mitose, Cdk1 devient inactive.
Les inhibiteurs physiologiques des Cdk (CKI)
L’activité des complexes cycline:Cdk est contrôlée par les CKI (cyclin-dependent kinase inhibitors), appartenant aux familles INK4 et Cip/Kip [9]. Les membres de la famille INK4 (p16INK4a, p15INK4b, p18INK4c et p19INK4d) contiennent des motifs ankyrine répétés. Ils se lient spécifiquement aux kinases Cdk4 et Cdk6, qu’ils inhibent en empêchant la liaison de la cycline D (Figure 2). La p16INK4a et la p15INK4b ont donc un rôle important en empêchant le déclenchement de la prolifération cellulaire, et sont de ce fait des gènes suppresseurs de tumeurs, fréquemment altérés dans les tumeurs. Notons au passage que le locus INK4a/ARF (alternative reading frame) contient deux gènes qui se chevauchent du fait d’un épissage alternatif. Les exons 1α, 2 et 3 codent pour le gène p14INK4a, alors que les exons 1β, 2 et 3 codent pour le gène p14ARF (p19ARF chez les rongeurs), qui est un gène suppresseur de tumeur qui agit négativement sur l’oncoprotéine Mdm2, impliquée dans la régulation négative de la protéine suppresseur de tumeur p53 (voir Figure 4, C4-5).
Les membres de la famille Cip/Kip (p21Waf1/Cip1/Sid1, p27Kip1 et p57kip2) se lient aux complexes cycline:Cdk en formant des hétérotrimères (Figure 2). La p27KIp1 est un gène suppresseur de tumeur bien établi, qui empêche la progression cellulaire en phase S en bloquant les complexes cycline E:Cdk2 et cycline A:Cdk2. La p21Waf1/Cip1/Sid1 a une spécificité plus large, puisqu’elle inhibe l’activité des complexes cycline E/A:Cdk2 et cycline B:Cdk1. Le gène p21Waf1/Cip1/Sid1 n’est généralement pas muté dans les tumeurs. La p21Waf1/Cip1/Sid1 est l’un des principaux médiateurs de l’arrêt du cycle cellulaire en réponse à divers stress cellulaires: en particulier, son promoteur est activé par la p53 (Figure 3, B1; Figure 4, D5). Dans certaines conditions, les protéines Cip/Kip pourraient également activer la progression du cycle cellulaire en facilitant la terminaison de la phase G1 et le début de la phase S. Cet effet s’expliquerait par la fixation des Cip/Kip aux complexes cycline D:Cdk4-6 qu’elles activeraient, et par libération et activation des complexes cycline E/A:Cdk2.
Le point de restriction (restriction point)
Le point de restriction, qui engage irréversiblement la cellule dans le cycle cellulaire est, schématiquement, contrôlé par la voie pRb-E2F, dont les anomalies sont fréquentes dans les cancers.
Les protéines de la famille pRb (pRb, pour retinoblastoma protein, p130 et p107), en se fixant aux protéines E2F, bloquent d’une façon générale leur activité transcriptionnelle (Figure 3, C4) dans les cellules quiescentes ou en début de phase G1. Cette inhibition de la transcription s’exerce également sous l’action du recrutement, par les protéines pRb, de désacétylases des histones (HDAC pour histone deacetylases), qui ont pour effet de compacter la chromatine et de la rendre inaccessible aux facteurs de transcription, en particulier E2F et DP. Dans les cellules quiescentes, les complexes E2F:DP1 sont inactivés par leur liaison à p130, celle-ci se trouvant substituée par pRb durant la phase G1. Pour assurer la transition G1→S, les complexes cycline D:Cdk4-6 phosphorylent pRb, ce qui entraîne un blocage de l’interaction pRb-E2F et permet à E2F d’activer l’expression des gènes codant pour les protéines essentielles à l’entrée en phase S et à la réplication de l’ADN. À l’inverse, les CKI maintiennent pRb dans son état répresseur, hypophosphorylé, en bloquant l’activité des kinases Cdk4 ou Cdk6 (Figure 3, B2-3). La terminaison de la phase S nécessite l’intervention de Cdk2 qui dissocie E2F de ses sites de liaison sur l’ADN.
La famille E2F est composée de 6 membres, activés par hétérodimérisation avec les protéines DP (DP1 ou DP2). Certains d’entre eux, les facteurs E2F4, E2F5 et surtout E2F6, pourraient avoir un effet régulateur négatif sur la transcription des gènes dépendant des facteurs E2F1-3:DP, par un effet compétitif de liaison avec les facteurs DP sur les mêmes promoteurs. Les gènes sous le contrôle des facteurs E2F:DP sont multiples [10]. Dans le groupe des gènes impliqués dans le cycle cellulaire, figurent les cyclines A et E, Cdk2 et Cdk1, Cdc25A, mais aussi pRb et p107. Les facteurs E2F activent également la transcription de gènes codant pour des protéines de réplication (Orc1, Mcm, Cdc6, Rfc 2-4, PCNA pour proliferating cell nuclear antigen), des enzymes nécessaires à la synthèse de l’ADN (ADN polymérase α, thymidylate kinase, dihydrofolate réductase), les systèmes de réparation de l’ADN (UDG, MutS, MutL, Rad51, DNA-PK, BRCA1), différents effecteurs des points de surveillance (p53, Chk1) et l’oncogène Myc (Figure 3, C4).
(→) m/s 2001, n°11, p. 1226
Une autre voie impliquée dans la prolifération cellulaire est celle de l’oncogène Myc, dont l’expression est ellemême étroitement liée à la prolifération cellulaire [11]. Myc est une protéine labile qui se fixe à son partenaire Max, une protéine stable et dépourvue d’activité transcriptionnelle propre. L’hétérodimère Myc:Max active la transcription en se fixant spécifiquement sur des séquences consensus de type boîtes E (E-boxes) (CANNTG). Parmi les gènes activés par Myc se trouvent ceux des cyclines D et E et de la phosphatase Cdc25A (Figure 3, A5) [12], qui toutes trois ont des rôles clé dans la transition G0→G1. Myc activerait également la transcription des gènes codant pour la protéine Cdk4 et les facteurs E2F (E2F2, E2F3 et Id2), et réprimerait celle du gène suppresseur de tumeur p21 [12].
Le TGFβ entraîne généralement l’arrêt de la prolifération celluaire, et la voie TGFβ est communément altérée dans les cancers. Le TGFβ produit cet effet à plusieurs niveaux: en réprimant l’expression de Myc, et en induisant l’expression de p15INK4b et de p21Waf1/Cip1 (dans certaines cellules).
L’induction de l’expression de la cycline E par E2F et Myc et, par voie de conséquence, l’activation de la kinase Cdk2 entraînent la phosphorylation de pRb (Figure 3, B4) et p27Kip1 (Figure 3, C1). La phosphorylation, et donc l’inactivation de pRb, crée une boucle rétro-activatrice qui libère l’activité d’E2F et contribue à l’irréversibilité de la transition G1→S, ce qui définit le point de restriction.
Pendant les phases S et G2, les kinases dépendantes des cyclines A et B (Cdk1 et Cdk2 sous forme de complexes avec leurs cyclines correspondantes) maintiennent la phosphorylation et l’inactivation de pRb, jusqu’au moment où les cellules sortent de mitose. La phosphorylation de l’inhibiteur de Cdk p27Kip1 par le complexe cycline E:Cdk2 conduit à sa dégradation (Figure 3, C1). Par ailleurs, ce même complexe active la synthèse d’ADN en phosphorylant Cdc45 (Figure 3, C1), et augmente la synthèse des histones en phosphorylant le facteur de transcription p220/NPAT (Figure 3, D1). L’activation du complexe cycline E:Cdk2 induit également la duplication des centrosomes en phosphorylant la nucléophosmine et la kinase MpS1.
Régulation moléculaire des points de surveillance du cycle cellulaire
L’identification des points de surveillance du cycle cellulaire (cell cycle checkpoints) a été l’une des contributions qui valut le prix Nobel à Hartwell, Nurse et Hunt. Ces points de surveillance assurent le contrôle de qualité du cycle cellulaire, et en particulier de la fidélité de la duplication du matériel génétique (→) [13]. En cas d’anomalies, ces mécanismes de surveillance stoppent la progression du cycle cellulaire et activent des processus de réparation ou de mort cellulaire par apoptose. Schématiquement, ce système de régulation est organisé en trois niveaux, avec des détecteurs (sensors), qui sont sensibles aux anomalies de l’ADN, aux défauts du cytosquelette ou aux perturbations métaboliques, des messagers (transducers), qui sont généralement des protéine kinases et des molécules régulatrices fonctionnant comme des adaptateurs et, enfin, des effecteurs (effectors), qui modifient directement les molécules impliquées dans le cycle cellulaire, la réplication et la réparation de l’ADN, ou l’apoptose. Les détecteurs ont été récemment revus par Zhou et Elledge [13] et Durocher et Jackon [14], et ne seront pas discutés dans cet article. Celle-ci se concentrera sur les messagers et les effecteurs. Les points de surveillance G1 et G2 sont les mieux compris au niveau moléculaire; quant au point de contrôle S, les éléments moléculaires sont en cours d’identification.
Les kinases dépendantes des phosphatidyl-inositols (PI3K-related kinases)
Cette famille de kinases est chargée de la coordination du cycle cellulaire avec le métabolisme de l’ADN et de la cellule [14]. Trois de ces kinases sont directement activées en présence de coupures de l’ADN: la kinase ATM (ataxia-telangiectasia mutated), la kinase ATR (ataxia-telangiectasia-like and Rad3 homolog) et la kinase DNA-PK (DNA-dependent protein kinase). Leur activation est impliquée dans l’arrêt du cycle permettant aux systèmes de réparation de travailler. Elles pourraient également activer ces systèmes de réparation et contrôler les voies de l’apoptose.
La kinase ATM est inactivée par mutation de son gène chez les malades atteints d’ataxie-télangiectasie, maladie autosomique récessive dont la caractéristique cellulaire est une déficience des points de surveillance du cycle cellulaire [15]. Ainsi, malgré la présence de coupures dans l’ADN, les cellules de patients atteints d’ataxie-télangiectasie continuent de proliférer et de synthétiser leur ADN, sans s’arrêter ni en phase S, ni en phase G2, c’est-à-dire sans laisser au système de réparation de l’ADN le temps d’agir. Il en résulte une hypersensibilité aux radiations ionisantes et aux agents inducteurs de lésions de l’ADN.
La kinase ATR étant essentielle à la survie cellulaire, aucune tumeur connue impliquant ATR n’a pu être détectée. Néanmoins, des travaux récents démontrent l’importance de la kinase ATR dans le signalement des anomalies au niveau des fourches de réplication, qui peuvent être observées au cours des lésions induites par les rayons ultraviolets ou les camptothécines, inhibiteurs des topo-isomérases I utilisés en chimiothérapie anticancéreuse, produisant des coupures double-brin de l’ADN au niveau des fourches de réplication [16].
La kinase DNA-PK est non seulement impliquée dans les points de surveillance du cycle cellulaire, mais aussi dans les recombinaisons non homologues de l’ADN. Elle est activée par les coupures double-brin de l’ADN [14]. Les anomalies de DNA-PK produisent des syndromes d’immunodéficience par défaut de recombinaison V(D)J des gènes des immunoglobulines G et du récepteur des lymphocytes T, et anomalies de maturation des cellules T.
Le rôle central des kinases ATM et ATR, et peut-être de la DNA-PK, dans les points de surveillance du cycle cellulaire est attesté par l’activité régulatrice de ces protéine kinases sur des protéines clé effectrices de la surveillance cellulaire telles que p53, Mdm2, le complexe cohésine, les kinases Chk1 et Chk2 (Figure 3, A1-2-3; Figure 4, A3-4) et différents détecteurs (sensors) tels que NBS1, BRCA1, hRad9 et hRad17.
Les protéine kinases Chk1 et Chk2
Chk1 et Chk2 (Figure 4, A1 et A2) sont deux sérine-thréonine kinases activées par ATM et ATR. Chk2 est exprimée indépendamment du cycle et de la prolifération cellulaire, alors que l’expression de Chk1 suit le cycle cellulaire, et est maximale pendant les phases S et G2. Chk2 est principalement activée par ATM, alors que Chk1 est principalement activée par ATR.
Il semble donc exister deux voies parallèles dans les cellules de mammifères: la voie ATR-Chk1 et la voie ATMChk2. La voie ATR-Chk1 répond principalement aux blocages des fourches de réplication et aux lésions produites par les rayons ultraviolets (Figure 4, A3), alors que la voie ATM-Chk2 répond aux cassures double-brin de l’ADN (Figure 4, A4). Nous avons récemment montré que l’inactivation de Chk2 par l’expression d’un oligonucléotide antisens diminue le ralentissement des phases S et G2 induit par le traitement des cellules par la camptothécine [17]. Cela souligne l’importance de Chk2 dans les mécanismes d’arrêt du cycle cellulaire mis en jeu lors des chimiothérapies.
Chk1 et Chk2 ont des substrats communs (p53 [Figure 4, B4], Cdc25 [Figure 4, C1] et BRCA1), et pourraient donc posséder des fonctions partiellement redondantes destinées à assurer la solidité du point de surveillance du cycle cellulaire. Chk2 semble jouer un rôle majeur en permettant la persistance de l’arrêt en G2, alors que Chk1 semble plus spécifiquement impliquée dans l’induction de l’arrêt en G2 [18].
La phosphatase Cdc25A et la voie p53-p21 dans le point de surveillance G1-S
Deux mécanismes agissent sur le point de contrôle G1-S [4] (Figure 3). L’un est rapide, et implique une cascade de kinases (ATM/ATR→Chk2) dont l’activation conduit à la dégradation, et donc à l’inactivation, de la phosphatase Cdc25A. Cela empêche l’activation des complexes cycline D:Cdk4/6 et cycline E/A:Cdk2 (Figure 3, A4).
Le second mécanisme est plus lent, car il implique l’activation de la protéine p53 et nécessite que soient exprimés les gènes activés par p53 (Figure 3, B1). Dans des conditions normales, p53 a une demi-vie très courte, et n’est présente qu’en faible quantité. Diverses perturbations (lésions de l’ADN, hypoxie ou surexpression d’oncogènes) augmentent les niveaux cellulaires de p53 (Figure 4, C4-5), principalement par sa phosphorylation sur la sérine 15 (par les kinases ATM et ATR, Figure 4, BC4) et sur la sérine 20 (par la kinase Chk2, mais aussi par les kinases Chk1 et Plk3) (Figure 4, B-C4-5). En effet, la phosphorylation de p53 réduit sa fixation de Mdm2, ce qui diminue son ubiquitinylation (sous le contrôle de Mdm2) et empêche ainsi sa dégradation dans le protéasome [19] (Figure 4, C4). La stabilisation de p53 induit l’expression de p21Waf1/Cip1 (Figure 4, C3) (inhibiteur des complexes cycline E/A:Cdk2 et cycline B:Cdk1), qui s’accompagne de l’arrêt du cycle cellulaire, et celle de gènes impliqués dans l’apoptose (Bax, DR5, Noxa, Fas). La protéine p53 induit également l’expression de Mdm2, ce qui produit un effet de rétrocontrôle négatif (feed-back loop) (Figure 4, D4-5). Lorsque la protéine p53 est déficiente, l’arrêt en G1 est atténué et les cellules entrent en phase S malgré la présence d’anomalies de l’ADN [20]. La protéine p14ARF est le produit d’un gène suppresseur de tumeurs (ARF) qui inactive Mdm2 en induisant sa translocation vers le nucléole, inhibe son activité ubiquitine ligase et augmente l’acétylation (donc la stabilisation) de p53 en bloquant l’effet inhibiteur de Mdm2 sur l’acétylase p300/CBP. La protéine ARF serait également induite par les radiations ionisantes, ce qui aurait pour effet de contribuer indirectement à l’activation de p53. L’importance de ARF comme régulateur positif de la voie p53 est attestée par le fait que les souris knock-out pour le gène ARF ont la même prédisposition aux tumeurs que les souris knock-out pour le gène de p53.
La phosphatase Cdc25C et la protéine p53 dans le point de surveillance G2-M
Le parallèle est remarquable avec le point de contrôle G1-S: deux types de réponses, rapide (par phosphorylation) ou lente (par contrôle transcriptionnel impliquant p53), assurent la surveillance de la transition G2→M.
Les kinases Chk1 et Chk2 jouent un rôle prédominant dans la réponse rapide, en phosphorylant Cdc25C sur la sérine 216 (Figure 4, C1). Cette phosphorylation crée un site d’attachement pour la protéine 14-3-3σ (Figure 4, C-D1), ce qui entraîne la séquestration de Cdc25C dans le cytoplasme, à distance de son substrat nucléaire, Cdk1. Des déficiences génétiques de 14.3.3 ont été impliquées dans les anomalies du point de surveillance G2 dans les cancers pulmonaires à petites cellules. Récemment, une nouvelle kinase de la famille des Polo kinases, Plk3, a été identifiée comme ayant les mêmes effets que Chk1 et Chk2 [21] (Figure 4, B-C1). Les lésions de l’ADN peuvent également inactiver Cdc25C en inhibant l’action de la Polo kinase Plk1, nécessaire à son activation (Figure 4, C1). L’inactivation de Cdk1 par les lésions de l’ADN peut aussi s’exercer via une hyperphosphorylation de son résidu Tyr15, par activation de Wee1 [22]. La réponse rapide peut aussi être due à une séquestration des complexes cycline B:Cdk1 dans le cytoplasme de cellules portant des lésions de l’ADN. Cela a pour effet d’empêcher la phosphorylation des substrats nucléaires impliqués dans la transition G2→M. La protéine p53 n’est pas nécessaire pour la décision d’arrêt en G2, mais elle joue un rôle dans sa persistance en bloquant l’activité du complexe kinase cycline B:Cdk1 [5]. Le contrôle qu’elle exerce emprunte diverses voies moléculaires (Figure 4, C3): l’induction de p21Waf1/Cip1 inhibe directement la kinase Cdk1, mais bloque également CAK, qui normalement active Cdk1 par phosphorylation de sa thréonine 161 ; l’induction de Gadd45 bloque elle aussi l’activité de la kinase Cdk1; l’induction de 14-3-3σ provoque la séquestration de la cycline B1 et de sa kinase Cdk1, ainsi que de Cdc25C dans le cytoplasme; la protéine p53 peut aussi réprimer l’expression des gènes Cdk1 et de la cycline B1 dont les promoteurs contiennent un site p53 (Figure 4, C5). Le nombre des voies moléculaires qui contrôlent le complexe cycline B1:Cdk1 est donc remarquable, reflétant l’importance d’une telle régulation.
Importance des altérations des composantes du cycle cellulaire et des points de surveillance dans les tumeurs humaines et expérimentales
Une prolifération sans frein et une résistance à la mort cellulaire par apoptose sont deux mécanismes fondamentaux de l’oncogenèse.
L’exemple des virus oncogènes : activation des kinases dépendantes des cyclines et inactivation des points de surveillance du cycle cellulaire
L’inactivation par des protéines virales des voies pRb et p53 représente l’un des mécanismes permettant la réplication virale, par induction de la division de la cellule hôte et blocage de sa mort par apoptose. Par exemple, le virus du papillome humain (HPV), responsable de carcinomes du col utérin, contient deux gènes essentiels, E6 et E7. La protéine E6 se lie à p53 et induit sa dégradation, tandis que la protéine E7 se lie à pRb et conduit à sa dégradation (Figure 3, B5). De plus, E7 inactive les CKI (cycline kinase inhibitors) tels que p21Waf1/Cip1. Dans le cas du virus SV40, la même protéine, l’antigène grand T, inactive à la fois p53 et pRb (Figure 3, B5).
Certains virus oncogènes peuvent activer directement les complexes cyclines:kinases de la phase G1, indépendamment de la protéine p53. Ainsi, le virus du sarcome de Kaposi (KSHV/HHV8) code pour une protéine homologue de la cycline D2 qui forme un complexe actif avec la protéine kinase Cdk6 (Figure 3, B1-2). Ce complexe induit également la dégradation de p27Kip1, ce qui contribue à l’activation des complexes cycline D:Cdk4/6.
Activation des voies moléculaires de la transition G1→S dans les cancers
Dans les modèles animaux, l’inactivation ou les mutations de pRb sont oncogéniques. Dans bon nombre de cancers humains, les cyclines D et leurs kinases associées (Cdk4, Cdk6) sont activées directement, ou indirectement par l’intermédiaire de déficiences en CKI [23]. Par exemple, la cycline D1 est surexprimée dans 70 % des lymphomes du manteau du fait d’une translocation t(11;14). La cycline E est fréquemment amplifiée dans les tumeurs humaines, et une élévation de la cycline E est corrélée avec des taux faibles de p27Kip1 (voir Figure 3, C1-2) et un mauvais pronostic chez les jeunes patientes atteintes de cancer du sein. Le facteur p27Kip1 est fréquemment sous-exprimé dans différents carcinomes (sein, côlon, estomac, poumons et prostate). D’autres anomalies génétiques affectent les protéines Cdk4, Cdk2, cycline E, ainsi que les CKI p15INK4B, p16INK4A et p57Kip2. Récemment, l’inactivation d’un nouvel inhibiteur du cycle cellulaire, CABLES, a été identifiée dans 50 % à 60 % des cancers du côlon, de la tête et du cou.
L’oncogène Myc est fréquemment amplifié dans les cancers [12, 24]. Outre des mécanismes moléculaires d’activation transcriptionnelle impliqués dans la transition G0→G1 et le point de restriction, Myc s’oppose à l’activité antiproliférative de TGFβ. Cet antagonisme peut s’expliquer par une liaison de Myc aux complexes transcriptionnels Smad/Sp1, impliqués dans la voie de signalisation du TGFβ, ainsi que par une répression directe des promoteurs des gènes p15INK4b et p21Waf1/Cip1.
Déficience des points de surveillance du cycle cellulaire dans les cancers
Le gène suppresseur de tumeur p53 est muté dans environ 50 % des cancers. En fait, les anomalies de la voie p53 sont fréquentes, même en l’absence de mutation de p53: c’est le cas des amplifications de Mdm2 (qui ont pour effet d’augmenter la dégradation de p53, Figure 4, B-C4) dans les ostéosarcomes. Les patients porteurs de mutations héréditaires de p53 (environ 80 % des sujets atteints du syndrome de Li-Fraumeni (→), une forme héréditaire de cancers touchant l’enfant et l’adulte jeune) présentent une prédisposition aux cancers, avec une incidence élevée de sarcomes, de carcinomes mammaires, cérébraux et de la surrénale, ainsi qu’une fréquence anormale de leucémies [25]. Récemment, des mutations de Chk2 ont été identifiées dans un sousgroupe de patients atteints d’un syndrome de Li- Fraumeni, mais dont la p53 est normale [26]. Cela souligne l’importance de Chk2 comme gène suppresseur de tumeurs, et de p53 comme substrat de Chk2 (phosphorylation sur la sérine 20) (Figure 4, B-C4-5).
Les mutations de la protéine kinase ATM sont responsables des syndromes d’ataxie-télangiectasie, qui prédisposent aux lymphomes et aux leucémies lymphoïdes chroniques (B ou T) [15]. Lorsque le gène ATM est normal (AT-like disorders), les mutations se situent au niveau de Mre11, un des éléments du complexe MRN (Mre11/Rad50/NBS1) impliqué dans la réparation des coupures double-brin de l’ADN. Les mutations de NBS1 sont quant à elle associées au syndrome de Nimègue (NBS pour Nijmegen breakage syndrome). Le groupement de ces anomalies moléculaires sous un syndrome commun paraît logique si l’on considère que la protéine ATM contrôle l’activité du complexe MRN en phosphorylant NBS1 (pour revue voir [27]).
Les mutations de BRCA1, qui est aussi un des gènes suppresseurs de tumeurs agissant sur les points de surveillance du cycle cellulaire, prédisposent aux cancers de l’ovaire et du sein (pour revue voir [27]).
Agents thérapeutiques ciblant les régulateurs du cycle cellulaire
L’objectif thérapeutique est de bloquer les voies moléculaires poussant les cellules tumorales dans le cycle cellulaire (inhibiteurs des Cdk ou des cyclines), ou d’agir au niveau des points de surveillance.
Les complexes cycline:Cdk étant suractivés dans la majorité des cancers, un des principes du développement des inhibiteurs de Cdk repose sur le fait que la prolifération et la survie des cellules tumorales dépendent de la suractivité des Cdk. L’adaptabilité des cellules normales permettrait d’arrêter temporairement leur prolifération, alors que la survie des cellules tumorales dépend des niveaux élevés de complexes cycline:Cdk. Il semble logique de développer des inhibiteurs sélectifs des Cdk de la phase G1-S, c’est-à-dire Cdk2/4/6, qui sont plus fréquemment suractivées dans les cancers que la Cdk1. Néanmoins, il a été observé que l’un des inhibiteurs de l’apoptose (IAP), la survivine, doit être phosphorylée par le complexe cycline B:Cdk1 pour pouvoir se lier à la caspase 9 et exercer son activité anti-apoptotique. Ces données suggèrent que l’inhibition des Cdk 2/4/6 pourrait avoir des effets cytotoxiques plus complexes, d’où l’intérêt de développer aussi des inhibiteurs de la Cdk1. Enfin, le fait que des inhibiteurs de Cdk (le flavopiridol, la roscovitine, les paullones) présentent une activité antitumorale constitue une justification empirique pour le développement des inhibiteurs du cycle cellulaire en oncologie médicale.
(→) m/s 2000, n°3, p. 425
En ce qui concerne les inhibiteurs des points de surveillance du cycle cellulaire, deux approches sont possibles. La première est de bloquer sélectivement un des points de surveillance, de façon à sensibiliser les tumeurs aux agents thérapeutiques classiques qui produisent des lésions de l’ADN. En effet, les déficiences de certains points de surveillance du cycle cellulaire (protéines p53, ATM ou Chk2) dans les cellules cancéreuses, à l’origine de l’incapacité de ces cellules d’arrêter leur progression dans le cycle, empêcherait la réparation des lésions de l’ADN et conduirait à leur mort cellulaire sélective. En revanche, dans les cellules normales, qui possèdent toute une gamme de points de contrôle, le blocage d’une voie de réparation ou d’un point de contrôle est compensé par des voies alternatives. L’autre approche est de rétablir les points de surveillance. Par exemple, les cellules tumorales dont la protéine p53 est déficiente meurent après complémentation de la p53 (voir plus loin).
Inhibiteurs des Cdk
Inhibiteurs aromatiques de la liaison de l’ATP aux Cdk
Ce sont généralement de petites molécules ayant un spectre d’action assez large sur les différentes Cdk. Des études cristallographiques ont démontré que ces inhibiteurs bloquent le site de liaison de l’ATP aux Cdk, et donc leur activation (pour revue, voir [1, 28–30]). Parmi ces inhibiteurs, le flavopiridol (Figure 5) est le plus avancé dans son développement clinique [31]. Cette molécule possède un large spectre d’activité sur les différentes Cdk (IC [concentration inhibitrice] 50 ≈ 0,1 μm pour les Cdk 1, 2 et 4, et 0,3 μm pour le complexe cycline H:Cdk7 [CAK]). Il provoque un arrêt en G1 et G2, et induit l’apoptose. L’arrêt en G1/ G2 relève d’au moins trois mécanismes : une inhibition directe des Cdk, une inactivation indirecte des Cdk 1, 2 et 4 par inhibition de la cycline H:Cdk7 (CAK) (voir Figure 3, A3) et une diminution de l’expression de la cycline D qui pourrait être due à une liaison du flavopiridol à l’ADN. Cliniquement, les concentrations plasmatiques de flavopiridol sont compatibles avec une activité sur les Cdk (0,3-0,5 μM). L’induction de l’apoptose est moins bien comprise. Elle pourrait être liée à une inactivation de la survivine, dont la liaison aux caspases dépendrait de sa phosphorylation par les Cdk. Le flavopiridol a une action synergique avec d’autres agents antitumoraux classiques. Des dérivés plus sélectifs et plus actifs sont en cours de développement, en particulier par Bristol Myers Squibb et Mitotix.
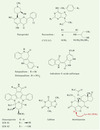 |
Figure 5. Inhibiteurs des Cdk et des points de surveillance du cycle cellulaire. |
La roscovitine (Figure 5) et l’olomoucine sont des dérivés puriques relativement peu puissants (IC50 de l’ordre du micromolaire sur Cdk4 ou Cdk6). Des dérivés plus actifs et plus spécifiques ont été obtenus par modélisation moléculaire et criblage de banques combinatoires. C’est le cas du CVT-313 (Figure 5) et du purvanolol, dont les substitutions sur le cycle purine augmentent l’affinité pour les Cdk. Ces dérivés sont plus spécifiques de Cdk1 que de Cdk4.
La kenpaullone appartient au groupe des paullones, dont la structure chimique n’a pas de similarité évidente avec l’ATP (Figure 5). Ces molécules ont été découvertes au National Cancer Institute par l’analyse COMPARE, fruit du travail de Ken Paull (d’où leur nom), en utilisant le flavopiridol comme référence. La kenpaullone inhibe Cdk1, Cdk2 et Cdk5, mais est beaucoup moins active contre Cdk4. Un de ses dérivés, l’alsterpaullone (Figure 5) est nettement plus actif in vitro sur la Cdk1 (≈10 fois) et dans des tests de cytotoxicité (IC50 ≈ 0,2 μM).
Récemment, l’indirubine (Figure 5), utilisée empiriquement en Chine pour traiter les leucémies myéloïdes chroniques, a été identifiée comme étant un inhibiteur de cyclines:Cdk [32]. L’indirubine-3’-monoxime stoppe la prolifération de diverses lignées cellulaires en G2 et des cellules leucémiques Jurkat en G1. Les dérivés de l’indirubine ont des effets potentiellement complexes, puisque certains dérivés se lient à l’ADN et aux microtubules [32]. Une des limitations des inhibiteurs de Cdk est que leur spectre d’activité sur les différentes Cdk est relativement large. Il serait en effet plus logique de développer des inhibiteurs de Cdk4 ou Cdk6 agissant sélectivement sur la voie pRb, qui est la plus fréquemment déficiente dans les cancers. Une série de pyridopyrimidines a ainsi été développée [33]; parmi ces molécules, le PD-171851 agit sélectivement sur la Cdk4, avec une IC50 de 0,042 μM. Ce dérivé arrête en G1 les cellules dont la voie pRb est normale, alors que les cellules dont la voie pRb est déficiente continuent de progresser dans le cycle cellulaire. Agouron-Pfizer a également présenté des dérivés 4-aminothiazoles sélectifs pour la Cdk4: l’AG-12275 est environ 100 fois plus actif sur la cycline D3:Cdk4 que sur la cycline A:Cdk2 ou la cycline B:Cdk1. Il arrête les cellules normales en G1. D’autres inhibiteurs, en cours de développement ou inclus dans des brevets, n’ont pas été mentionnés ici. Des informations complémentaires sont disponibles dans la récente revue de Fry et Garrett [28].
Inhibiteurs peptidiques et peptidomimétiques des Cdk
Des études réalisées in vitro et in vivo démontrent que la surexpression de p16INK4a prévient la croissance tumorale. Des dérivés peptidiques mimant l’action des CKI (p16Ink4a, p21Cip1) ont quant à eux fait la preuve de leur activité dans des systèmes cellulaires in vitro (pour revue voir [31]). Néanmoins, ces composés sont expérimentaux, et leur couplage à une séquence peptidique de la protéine du gène Antennapedia de drosophile a été utilisé pour permettre leur pénétration cellulaire et démontrer la validité de cette approche. Par ailleurs, un polypeptide de 36 acides aminés (résidus 84-103 de p16INK4a) couplé à un peptide transporteur bloque Cdk4 et la prolifération cellulaire. Du fait que p16INK4a bloque sélectivement Cdk4 et Cdk6, mais pas les autres Cdk (Figure 2), il semble que le ciblage sélectif des protéine kinases Cdk4 et Cdk6 aurait pour effet de bloquer sélectivement la prolifération des cellules ayant une voie pRb normale. Lorsque les tumeurs contiennent une protéine pRb défectueuse, ces inhibiteurs pourraient aussi être utiles pour protéger les cellules normales de l’hôte vis-à-vis de thérapeutiques antitumorales agissant sur les cellules en cours de division cellulaire.
Inhibiteurs des point de surveillance du cycle cellulaire
L’UCN-01 et les indolocarbazoles
La 7-hydroxystaurosporine (UCN-01, Figure 5) est en essai clinique (phases II/III). L’UCN-01 fut isolé de cultures de fermentation de Streptomyces et initialement caractérisé comme un inhibiteur de la protéine kinase C (pour revue voir [1, 28–30]). L’UCN-01 est actif sélectivement contre les isozymes classiques de la PKC dépendant du Ca2+ (α,β ou γ) (avec une IC50 de 30 nM) et n’est pas actif contre la PKCξ. L’UCN-01 est plus sélectif que son stéréo-isomère l’UCN-02 ou que la staurosporine (Figure 5). Une différence importante avec les autres indolocarbazoles, tels la rébécamycine ou le NB-506, est que l’UCN-01 (comme la staurosporine) ne se fixe pas à l’ADN et n’a pas d’effet sur la topo-isomérase I.
Suivant la dose [29], l’UCN-01 peut abroger les points de surveillance du cycle cellulaire, arrêter la progression cellulaire en G1 ou induire l’apoptose. Ces deux derniers effets sont généralement observés à des concentrations supérieures à 0,5 μM. L’arrêt en G1 est corrélé avec une déphosphorylation de pRb, une inhibition de Cdk2 par déphosphorylation, une réduction des niveaux de cycline A et une activation de p21WAF1/Cip1. Il est également dû à une protéolyse de E2F1.
À des concentrations observées en clinique (< 0,3 μM), l’UCN-01 n’a qu’une activité antiproliférative modeste. Néanmoins, dans ces conditions, il potentialise remarquablement l’activité des chimiothérapies classiques qui produisent des lésions de l’ADN (comme la mitomycine C, le témolozomide, le cisplatine ou la camptothécine) ou interfèrent avec le métabolisme de l’ADN (5-fluoro-uracile ou méthotrexate). Cette synergie s’explique par une suppression des points de surveillance de G2 et de S, et est généralement plus importante pour les cellules présentant des anomalies de la voie p53.
Les cibles moléculaires de l’UCN-01 ont été clarifiées récemment. Pour le point de contrôle G2, la cible la plus vraisemblable est la kinase Chk1 [34]. De plus, l’UCN-01 pourrait aussi activer Cdc25C en inhibant Wee1 (Figure 4, D2). Nous avons également observé que l’UCN-01 peut bloquer l’activité de Chk2 dans des précipités de complexes immuns [18]. Il semble donc que l’effet suppresseur sur les points de surveillance du cycle cellulaire puisse être la résultante d’une inhibition de plusieurs kinases, en particulier Chk1, et possiblement Chk2 et Wee1. L’inhibition de Chk2 pourrait expliquer la suppression du point de contrôle de la phase S observé dans les cellules traitées par la camptothécine [35]. Un autre effet de l’UCN-01 en relation avec le point de contrôle S est l’inhibition de la phosphorylation de la protéine de réplication et de réparation RPA2 (replication protein A) dans les cellules traitées par la camptothécine [35]. Dans ces conditions, la kinase qui phosphoryle RPA2 est la DNA-PK [35], ce qui suggère soit que Chk1 ou Chk2 peuvent moduler la DNA-PK, soit que l’UCN-01 agit sur d’autres kinases qui n’ont pas encore été identifiées. Des observations récentes suggèrent également que l’UCN-01 potentialise l’activité du cisplatine en inhibant la réparation par excision de nucléotides, via son interférence dans les interactions entre XPA et ERCC1 (protéines de réparation par excision).
L’UCN-01 est en cours d’évaluation clinique, et les études de phase I ont révélé des limitations pharmacocinétiques car, à la différence des études animales, l’UCN-01 se lie fortement à la glycoprotéine acide α-1 et son élimination est très lente (environ 30 jours) [29]. Les essais cliniques de phase II/III associant l’UCN-01 et des agents qui produisent des lésions de l’ADN sont en cours.
Caféine
La caféine (Figure 5) est le plus ancien inhibiteur des points de surveillance du cycle cellulaire. Elle abroge le point de contrôle G2 (à des concentrations élevées, de l’ordre du millimolaire) en supprimant les phosphorylations inhibitrices sur les complexes cycline B:Cdk1 (pour revue voir [30]). Elle abroge aussi le point de contrôle G1 et réduit la stabilisation de p53 après irradiation. La caféine surmonte également le point de contrôle S et bloque la phosphorylation régulatrice de Chk1 dans les extraits de xénope et dans la levure S. pombe. Ces effets multiples ont récemment été attribués à l’inhibition directe de la protéine kinase ATM. La caféine inhibe également la kinase ATR, ainsi que rad3, l’homologue de ATM et ATR chez la levure S. pombe.
Wortmannine
La wortmannine (Figure 5) est un dérivé mycosique qui inhibe irréversiblement, à des concentrations relativement basses (1-20 nM), les protéine kinases de la famille PI3K (pour revue voir [30]). À des concentrations plus élevées (0,2-0,5 μM), la wortmannine inhibe également les protéine kinases DNA-PK et ATM, ainsi que rad3 et mTOR (mammalian target of rapamycin). À ces concentrations, la wortmannine n’a pas d’effet notable sur la protéine kinase C, les protéine kinases dépendant de l’AMPc ou du GMPc, les MAP kinases, la p70S6k ou la protéine kinase associée au récepteur du PDGF (platelet derived growth factor). Des expériences utilisant la wortmannine radiomarquée suggèrent que le groupement C-20 de la wortmannine forme un adduit covalent sur des résidus lysine essentiels du domaine phosphotransférase de PI3K (peutêtre la lysine 802) (Figure 5). La wortmannine bloque la stabilisation de p53 et le blocage de la réplication de l’ADN (point de contrôle S), comme on l’attendait d’un agent bloquant la kinase ATM. La wortmannine est un radiosensibilisateur puissant. Elle présente également un effet synergique avec le chlorambucil [36] et l’étoposide, de façon indépendante du statut p53. Les effets inhibiteurs de la wortmannine sur les kinases de la famille PI3K peuvent expliquer ses effets inhibiteurs sur la réparation des coupures double-brin de l’ADN, la suppression du point de contrôle de la phase S et l’inhibition de la phosphorylation de RPA2 en réponse aux coupures de l’ADN [35].
La wortmannine a une faible activité antitumorale dans les modèles animaux et sa réactivité chimique exclut son usage clinique. Néanmoins, il pourrait être intéressant de développer des agents qui inhiberaient sélectivement les kinases DNA-PK ou ATM.
Autres sites d’intervention
Les inhibiteurs des phosphatases Cdc25
La protéine Cdc25C est récemment apparue comme une nouvelle cible [37] du fait de sa surexpression dans une variété de cancers. La phosphatase Cdc25A est également une cible transcriptionnelle de l’oncogène c-myc (voir plus haut). L’élucidation de la structure cristalline du domaine catalytique des Cdc25 devrait faciliter le développement d’inhibiteurs.
Modulation du facteur de transcription et de prolifération cellulaire E2F-1
Des vecteurs adénoviraux ont démontré qu’il est possible de surexprimer E2F-1 et de déclencher l’apoptose par des voies dépendantes de p53, mais aussi par des voies indépendantes de p53 impliquant p73 (pour revue voir [30]). Les antagonistes de Cdk2 ont une activité cytotoxique spécifique pour les cellules qui ont une suractivité du facteur de transcription E2F-1. Cela peut s’expliquer par le fait que l’association entre une activation de E2F-1 et une inhibition de la Cdk2 est, dans les modèles de levure, « synthétiquement létale ».
Thérapie génique ciblant les cellules déficientes pour la p53
L’adénovirus du groupe Cdl1520 (Onyx-015) a été conçu pour se multiplier sélectivement dans les cellules déficientes pour p53. Cette sélectivité est liée au fait que le virus Onyx-015 n’exprime pas le gène viral E1B 55-kDa dont la fonction est de neutraliser p53 qui, normalement, bloque la réplication virale [38]. Bien que le virus Onyx-015 soit actif par lui-même dans des tumeurs ORL, les réponses sont généralement temporaires. Des essais cliniques de phase II combinant l’Onyx-015 avec du cisplatine et du 5-fluoro-uracile ont montré des régressions spectaculaires et parfois complètes [39]. Des données récentes suggèrent que l’Onyx-015 peut se répliquer et tuer des cellules dont la protéine p53 est normale, ce qui suggère une complexité plus grande qu’initialement envisagé. En particulier, une perte de p14ARF facilite la réplication de l’Onyx-015. l’Onyx-015 pourrait donc être utile dans des tumeurs avec des anomalies indirectes de la voie p53.
Ciblage de l’interface moléculaire p53-Mdm2
Mdm2 est l’antagoniste physiologique de p53 (Figure 4, C-D.4-5), comme en témoigne l’observation remarquable que le knock-out de MDM2 est mortel chez la souris, sauf si p53 est elle aussi inactivée (pour revue voir [19]). En conséquence, il a été proposé de développer des thérapeutiques anti-Mdm2 qui bloqueraient les interactions Mdm2-p53 dans les tumeurs exprimant une p53 normale. Il en résulterait donc une activation non contrôlée de p53 et la mort cellulaire par apoptose ou arrêt dans le cycle [20]. Des petits peptides qui se lient au site de fixation de Mdm2 dans la région N-terminale de p53 ont été identifiés. Ils bloquent la liaison de Mdm2 sans interférer avec les autres fonctions de p53. Récemment, une équipe de Novartis a découvert un octapeptide dérivé de p53 qui bloque l’interaction p53- mdm2 avec une affinité élevée in vitro, et qui est cytotoxique dans les tumeurs avec surexpression de Mdm2 [40]. Une autre façon d’activer p53 indirectement peut être d’utiliser un peptide dérivé de la région N-terminale de p14ARF, de façon à neutraliser Mdm2 (Figure 4, C-D4- 5) [41]. Finalement, Hybridon a récemment montré qu’il est possible d’inhiber l’expression de Mdm2 par des oligonucléotides antisens, de façon à activer la p53. Cette approche a fait la preuve de son efficacité par ellemême et en association avec l’adriamycine et la 10- hydroxycamptothécine.
Une nouvelle approche très prometteuse pour réactiver p53 dans les cellules contenant une p53 mutée est d’utiliser des substances qui restaurent l’activité fonctionnelle de la protéine p53 mutée. Des petites molécules ayant cette activité ont récemment été décrites (pour revue voir [42]). Il sera intéressant de tester leur activité en association avec des agents chromolithographiques classiques qui induisent l’apoptose de façon dépendante de p53.
Ciblage de Myc
Comme nous l’avons discuté précédemment (section II.3), c-Myc est un oncogène qui joue un rôle critique dans la transition G0→G1 et le « point de restriction » [12]. Des études récentes réalisées dans des modèles murins ont démontré la réversibilité du phénotype malin après suppression de l’expression de Myc [24]. Le blocage de Myc est donc une approche thérapeutique logique: récemment, Parodi et al. ont développé des polypeptides bloquant la liaison de Myc à l’ADN [43, 44]. Il est également concevable d’envisager le blocage de la formation des complexes Myc:Max (Figure 3, A5).
Conclusions
Il est aujourd’hui clair que de nombreux cancers comportent des anomalies du cycle cellulaire et de leurs points de surveillance: activation des protéine kinases dépendantes des cyclines (amplification des cyclines, des Cdk, diminution des CKI tels que p16INK4a…), déficiences des points de surveillance du cycle cellulaire (mutations de p53, des kinases ATM ou Chk2…). La possibilité de mesurer ces paramètres dans des tumeurs, en pratique oncologique, permet d’envisager l’utilisation ciblée de nouvelles thérapeutiques anticancéreuses. L’identification des mécanismes moléculaires formant l’infrastructure de ces voies de signalisation permet également d’envisager la réalisation d’essais pour développer et tester de nouvelles molécules inhibitrices. Le défi des années à venir est d’intégrer les voies de signalisation du cycle cellulaire et de leurs points de surveillance dans chaque tumeur, afin d’adapter les traitements à des situations moléculaires spécifiques correspondant à chaque patient.
Références
- Borgne A, Meijer L. Inhibiteurs chimiques des kinases dépendantes des cyclines: recherche et applications thérapeutiques potentielles. Med Sci 1999; 15: 496–503. [Google Scholar]
- Sherr CJ, DePinho RA. Cellular senescence: mitotic clock or culture shock? Cell 2000; 102: 407–10. [Google Scholar]
- Bartek J, Falck J, Lukas J. Chk2 kinase - a busy messenger. Nat Rev Mol Cell Biol 2001; 2: 877–86. [Google Scholar]
- Bartek J, Lukas J. Mammalian G1- and S-phase checkpoints in response to DNA damage. Curr Opin Cell Biol 2001; 13: 738–47. [Google Scholar]
- Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene 2001; 20: 1803–15. [Google Scholar]
- Kohn KW. Molecular interaction map of the mammalian cell cycle control and DNA repair systems. Mol Biol Cell 1999; 10: 2703–34. [Google Scholar]
- Bartek J, Lukas J. Cell cycle: order from destruction. Science 2001; 294: 66–7. [Google Scholar]
- Nilsson I, Hoffmann I. Cell cycle regulation by the Cdc25 phosphatase family. Prog Cell Cycle Res 2000; 4: 107–14. [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1- phase progression. Genes Dev 1999; 13: 1501–12. [Google Scholar]
- Ren B, Cam H, Takahashi Y, et al. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev 2002; 16: 245–56. [Google Scholar]
- Trumpp A, Refaeli Y, Oskarsson T, et al. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 2001; 414: 768–73. [Google Scholar]
- Grandori C, Cowley SM, James LP, Eisenman RN. The myc/max/mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol 2000; 16: 653–99. [Google Scholar]
- Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408: 433–9. [Google Scholar]
- Durocher D, Jackson SP. DNAPK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr Opin Cell Biol 2001; 13: 225–31. [Google Scholar]
- Shiloh Y, Kastan MB. ATM:genome stability, neuronal development, and cancer cross paths. Adv Cancer Res 2001; 83: 209–54. [Google Scholar]
- Pommier Y. Les ADN topoisomérases, gardes-barrières du génome et leur sabotage par les antibiotiques et anticancéreux. Med Sci 1994; 10: 953–5. [Google Scholar]
- Yu Q, Rose JH, Zhang H, Pommier Y. Antisense inhibition of Chk2/hCds1 expression attenuates DNA damage-induced S and G2 checkpoints and enhances apoptotic activity in HEK- 293 cells. FEBS Lett 2001; 505: 7–12. [Google Scholar]
- Yu Q, La Rose JH, Zhang H, Takemura H, Kohn KW, Pommier Y. UCN-01 inhibits p53 up-regulation and abrogates gammaradiation- induced G(2)-M checkpoint independently of p53 by targeting both of the checkpoint kinases, Chk2 and Chk1. Cancer Res 2002; 62: 5743–8. [Google Scholar]
- Daujat S, Neel H, Piette J. MDM2: life without p53. Trends Genet 2001; 17: 459–64. [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997; 88: 323–31. [Google Scholar]
- Xie S, Wu H, Wang Q, et al. Plk3 Functionally links DNA damage to cell cycle arrest and apoptosis at least in part via the p53 pathway. J Biol Chem 2001; 276: 43305–12. [Google Scholar]
- Fletcher L, Cheng Y, Muschel RJ. Abolishment of the Tyr-15 inhibitory phosphorylation site on cdc2 reduces the radiation-induced G(2) delay, revealing a potential checkpoint in early mitosis. Cancer Res 2002; 62: 241–50. [Google Scholar]
- Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 2001; 1: 222–31. [Google Scholar]
- Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell 1999; 4: 199–207. [Google Scholar]
- Lane DP. Exploiting the p53 pathway for cancer diagnosis and therapy. Br J Cancer 1999; 80 (suppl 1): 1–5. [Google Scholar]
- Lee SB, Kim SH, Bell DW, et al. Destabilization of CHK2 by a missense mutation associated with Li-Fraumeni syndrome. Cancer Res 2001; 61: 8062–7. [Google Scholar]
- Wang JYJ. New link in a web of human genes. Nature 2000; 405: 404–5. [Google Scholar]
- Fry DW, Garrett MD. Inhibitors of cyclin-dependent kinases as therapeutic agents for the treatment of cancer. Curr Opin Onc End Met Invest New Drugs 2000; 2: 40–59. [Google Scholar]
- Senderowicz AM. Small molecule modulators of cyclin-dependent kinases for cancer therapy. Oncogene 2000; 19: 6600–6. [Google Scholar]
- Pommier Y, Yu Q, Kohn KW. Novel targets in the cell cycle and cell cycle checkpoints. In : Baguley BC, Kerr DJ, eds. Anticancer drug development. San Diego : Academic Press, 2002 : 13–30. [Google Scholar]
- Senderowicz AM, Sausville EA. Preclinical and clinical development of cyclindependent kinase modulators. J Natl Cancer Inst 2000; 92: 376–87. [Google Scholar]
- Hoessel R, Leclerc S, Endicott JA, et al. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat Cell Biol 1999; 1: 60–7. [Google Scholar]
- Boschelli DH, Dobrusin EM, Doherty AM, et al. Pyrido [2,3-D] pyrimidines and 4- aminopyrimidines as inhibitors of cellular proliferation. Warner- Lambert Co, 1998;: WO- 09833798. [Google Scholar]
- Graves PR, Yu L, Schwarz JK, et al. The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J Biol Chem 2000; 275: 5600–5. [Google Scholar]
- Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA: DNA-PK complexes. EMBO J 1999; 18: 1397–406. [Google Scholar]
- Christodoulopoulos G, Muller C, Salles B, Kazmi R, Panasci L. Potentiation of chlorambucil cytotoxicity in B-cell chronic lymphocytic leukemia by inhibition of DNA-dependent protein kinase activity using wortmannin. Cancer Res 1998; 58: 1789–92. [Google Scholar]
- Eckstein JW. Cdc25 as a potential target of anticancer agents. Invest New Drugs 2000; 18: 149–56. [Google Scholar]
- Heise C, Sampson-Johannes A, Williams A, McCormick F, Von Hoff DD, Kirn DH. Onyx- 015, an E1B gene-attenuated adenovirus, causes tumorspecific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med 1997; 3: 639–45. [Google Scholar]
- Khuri FR, Nemunaitis J, Ganly I, et al. A controlled trial of intratumoral ONYX- 015, a selectively-replicating adenovirus, in combination with cisplatin and 5- fluorouracil in patients with recurrent head and neck cancer. Nat Med 2000; 6: 879–85. [Google Scholar]
- Chene P, Fuchs J, Bohn J, Garcia-Echeverria C, Furet P, Fabbro D. A small synthetic peptide, which inhibits the p53-hdm2 interaction, stimulates the p53 pathway in tumour cell lines. J Mol Biol 2000; 299: 245–53. [Google Scholar]
- Midgley CA, Desterro JM, Saville MK, et al. An Nterminal p14ARF peptide blocks Mdm2-dependent ubiquitination in vitro and can activate p53 in vivo, Oncogene 2000; 19: 2312–23. [Google Scholar]
- Novak K. p53 restoration. Nat Rev Cancer 2002; 2: 159. [Google Scholar]
- Giorello L, Clerico L, Pescarolo MP, et al. Inhibition of cancer cell growth and c-Myc transcriptional activity by a c-Myc helix 1-type peptide fused to an internalization sequence. Cancer Res 1998; 58: 3654–9. [Google Scholar]
- Pescarolo MP, Bagnasco L, Malacarne D, et al. A retroinverso peptide homologous to helix 1 of c-Myc is a potent and specific inhibitor of proliferation in different cellular systems. Faseb J 2001; 15:31–3. [Google Scholar]
- Rothblum-Oviatt CJ, Ryan CE, Piwnica-Worms H. 14-3-3 binding regulates catalytic activity of human wee1 kinase. Cell Growth Differ 2001; 12: 581–9. [Google Scholar]
Liste des figures
|
Figure 1. Symboles et conventions utilisés [6]. (a) La double flèche indique que les protéines A et B se lient l’une à l’autre. Le point sur la ligne représente le complexe A:B. (b) Représentation de complexes multimoléculaires: x est [A:B] et y est [(A:B):C]. Cette notation peut être étendue à un nombre plus grand de molécules dans un complexe. (c) Modification covalente de la protéine A. La flèche indique que A peut exister à l’état phosphorylé. Le point représente la protéine A phosphorylée. (d) Clivage d’une liaison covalente: déphosphorylation de A par une phosphatase. (e) Conversion stoechiométrique de A à B. Dégradation de A (c’est-à-dire conversion de A en son produit de dégradation Ø). (f) Stimulation enzymatique d’une réaction. (g) Symbole général de stimulation. (h) Symbole général d’inhibition. (i) Symbole d’activation de la transcription. |
| Dans le texte | |
|
Figure 2. Associations moléculaires entre cyclines, Cdk et CKI. Il existe au moins neuf Cdk (cyclin-dependent kinases), les protéines Cdk 1 à 7 ayant été clairement impliquées dans le cycle cellulaire. Cdk7, Cdk8 et Cdk9 sont impliquées dans la transcription. Il existe au moins 15 cyclines (cyclines A à T) dont l’expression varie durant le cycle cellulaire. Les protéines Cdk sont activées par leur liaison aux cyclines et inactivées par leur liaison aux CKI. L’échelle à gauche représente les phases du cycle cellulaire durant lesquelles les complexes cyclines:Cdk sont actifs. (1) La cycline D (CycD) se lie à Cdk4 ou Cdk6 (le complexe bimoléculaire est représenté par le point noir, voir Figure 1). (2) La cycline E (CycE) se lie à Cdk2. (3) La cycline A (CycA) se lie à Cdk2 durant la phase S et à Cdk1 (appelée aussi Cdc2) en fin de phase S et durant la phase G2. (4) La cycline B (CycB) se lie à Cdk1. (5) Les CKI (cyclin-dependent kinase inhibitors) p16INK4a ou p15INK4b entrent en compétition avec la cycline D pour se fixer à Cdk4 ou Cdk6, ce qui empêche l’activation de ces kinases. (6) p21Waf1/Cip1 et p27kip1 se lient aux complexes cycline D:Cdk4 ou cycline D:Cdk6. Lorsque p21cip1 et p27kip1 sont en excès, il en résulte une inhibition. En revanche, de faibles concentrations de p21Waf1/Cip1 pourraient avoir un effet stimulant en empêchant la dégradation de la cycline D. (7) p27kip1 ou p21Waf1/Cip11 se lient aux complexes cycline E:Cdk2 ou cycline A:Cdk2 et inhibent leur activité kinase. (8) p21Waf1/Cip1 peut également se lier au complexe cycline B:Cdk1 et inhiber son activité. |
| Dans le texte | |
|
Figure 3. Interactions moléculaires de la transition G1→S et du contrôle du facteur de transcription E2F. Les symboles sont définis par Kohn [6]. (1) [coordonnées D3] La transition G1→S implique les gènes contrôlés par E2F et la cycline E. (2) [C5] Les gènes dépendants de E2F codent pour des protéines régulatrices telles que la cycline E et la protéine Myc, et des gènes dont les produits sont des enzymes impliquées dans la réplication (non incluses sur le schéma). Ces gènes sont activés par les complexes E2F1:DP1 (et d’autres hétérodimères E2F) et négativement réglés lorsque E2F est lié à pRb. Cette dernière possède donc un rôle majeur. (3) [B4] La liaison de pRb à l’hétérodimère d’E2F est inhibée lorsque pRb est phosphorylée. pRb est également inhibée par les protéines virales E1A et l’antigène grand T (SV40-T). (4) [B5] La dégradation de pRb est stimulée par la protéine E7 du virus du papillome. (5) [B4] pRb est phosphorylée par les complexes cycline D:Cdk4/6 et cycline E:Cdk2. Ces deux complexes kinases sont nécessaires pour phosphoryler pRb complètement et bloquer l’interaction entre pRb et E2F. (6)[B.2] Cdk4 et Cdk6 sont actives quand elles sont couplées à la cycline D. (7)[B2] Cdk4 et Cdk6 peuvent être phosphorylées à des sites qui stimulent ou bloquent leur activité. (8)[B2] Les CKI p15 ou p16 peuvent se fixer à Cdk4 ou Cdk6 et bloquer leur fixation de la cycline D. Cela a pour effet d’inactiver les kinases. (9)[A2] Le TGFβ active la transcription de p15 par la voie des Smad (non montré). (10)[B2] La protéine Cdk6 peut être inactivée par déplacement de la cycline D par la protéine virale KSHV/HHV8 du virus du sarcome de Kaposi. (11)[B2] Les hétérodimères cycline D:Cdk4/6 peuvent se lier à p21cip1 ou p27kip1, ce qui a pour effet de les stabiliser. (12)[B2] La cycline D a une demi-vie courte, d’environ 30 minutes. Elle est dégradée dans le protéasome après ubiquitinylation. (13)[B.2] La protéine p53 active p21cip1. (14)[B2] p21cip1 se fixe à PCNA, un co-facteur de l’ADN polymérase. (15)[C.2] p21cip1 ou p27kip1 se lient aux complexes cycline E:Cdk2 et bloquent leur activité kinase (par opposition aux complexes cycline D:Cdk4/6 (voir [11] ci-dessus). (16)[C2] p27kip1 est phosphorylée par la cycline E:Cdk2. p27kip1 est donc à la fois un substrat et un inhibiteur des complexes kinases cycline E. (17)[C1] p27kip1 est dégradée après phosphorylation. (18)[C2] La liaison de la protéine virale KSHV/HHV8 stimule la dégradation de p27kip1. (19)[C2-3] Les complexes cycline E:Cdk2 (et cycline D:Cdk4/6) sont phosphorylés au niveau de sites d’activation et d’inhibition. (20)[A3] Cdk2, Cdk4 et Cdk6 sont phosphorylées et activées par la cycline H:Cdk7 (encore connue sous le nom de CAK), qui est un composant du facteur de transcription TFIIH. (21)[B3] Cdk2, Cdk4 et Cdk6 sont activées par déphosphorylation par la phosphatase Cdc25A. (22)[A4] Cdc25A est activée par phosphorylation par Raf1 dans la voie de signalisation Ras. (23)[A5] La transcription du gène de Cdc25A est activée par les complexes c-Myc:Max. (24)[D2-3] Le complexe cycline A:Cdk2 phosphoryle E2F1 et DP1, ce qui produit la dissociation du complexe E2F1:DP1, avec pour effet d’arrêter l’activité de E2F en fin de phase S. Les conventions utilisées pour le choix des couleurs sont les suivantes; en rouge, modifications covalentes et voies de transcription; en vert, modifications enzymatiques; en bleu: effets stimulants ou inhibiteurs; en noir, liaisons intermoléculaires; lettres rouges: inputs et outputs. |
| Dans le texte | |
|
Figure 4. Interactions moléculaires de la transition G2→M. (1)[coordonnées E3] La transition G2→M, qui prépare à la mitose, est principalement contrôlée par le complexe kinase cycline B:Cdk1. (2)[D2] L’activité kinase de Cdk1 est contrôlée par des phosphorylations spécifiques. La phosphorylation de la thréonine 161 est nécessaire pour son activité, alors que la phosphorylation de la thréonine 14 et de la tyrosine 15 sont inhibitrices. (3)[D2] La thréonine 161 de Cdk1 est phosphorylée par la cycline H:Cdk7 (CAK) (voir Figure 3, phosphorylations stimulantes de Cdk2 et Cdk4/6). (4)[D2] La tyrosine kinase nucléaire Wee1 phosphoryle spécifiquement la tyrosine 15 de Cdk1 (et de Cdk2) pendant les phases S et G2, alors que la kinase cytoplasmique Myt1 a une double spécificité pour la thréonine 14 et la tyrosine15 de Cdk1. (5)[C2] Inversement, ces résidus sont déphosphorylés par la phosphatase Cdc25C. (6)[C1] Cdc25C peut être activée par phosphorylation par la Polo kinase Plk1 et par le complexe cycline B:Cdk1, ce qui produit une boucle de contrôle positif pour l’activation de ce complexe. (7)[C1] La phosphorylation de Cdc25C sur la sérine 216 stimule sa liaison à la protéine 14–3–3σ, avec pour résultat une inhibition de sa phosphatase. La liaison de 14–3–3σ à la kinase Weel stimule également l’activité de Wee1 (non montré) [45]. La protéine 14–3–3σ a donc au moins deux mécanismes régulateurs négatifs sur la progression G2→M. (8)[C1-2] La phosphorylation de Cdc25C sur la sérine 216 peut être catalysée par Chk1, Chk2 ou Plk3 (voir annotation 25 ci-dessous). (9)[B1] Chk1 et Chk2 sont activées par phosphorylation. (10)[B2-3] Chk1 semble être principalement phosphorylée par ATR, alors que Chk2 peut être phosphorylée par ATM (ou ATR). (11)[A3] ATR est activée lors de la détection de blocages des fourches de réplication de l’ADN produites par les ultraviolets (UV) ou la camptothécine (CPT). (12)[A4] ATM est activée par des coupures double-brin de l’ADN (DSB) produites par divers agents tels que les radiations ionisantes (IR) ou l’inhibiteur de topo-isomérase II, l’étoposide (VP16). (13)[A-B2-3] La caféine inhibe ATM et ATR. (14)[A2] L’UCN-01 inhibe une variété de protéine kinases, dont Chk1 et Chk2, mais aussi Wee1. Cet effet est indirect. (15)[B4] ATM peut phosphoryler p53 sur sa sérine 15, ce qui stabilise p53 en réduisant sa dégradation. (16)[B3-4] Chk1 et Chk2 phosphorylent p53 sur la sérine 20, ce qui a pour effet de bloquer la liaison de Mdm2. (17)[C5] Mdm2 est un antagoniste de p53, agissant d’une part en l’ubiquitinylant, ce qui déclenche sa dégradation, et d’autre part en bloquant son activité transcriptionnelle. (18)[D5] Mdm2 se fixe à p14Arf, ce qui a pour effet de diminuer la dégradation de p53 liée à la fixation de Mdm2. (19)[E4] Mdm2 se fixe à E2F1 et stimule sa dégradation. Cela bloquerait l’activation des gènes spécifiques de la phase S (voir Figure 3). (20)[D5] La protéine p53 active de nombreux gènes, y compris ceux de Mdm2, p21cip1, Gadd45, 14–3–3 et Bax. (21)[C5] La p53 inhibe d’autre gènes, comme ceux de la cycline B1 et de Cdk1. (22)[C2-3] p21cip1 inhibe plusieurs complexes cycline-kinase (voir Figure 2), comme la cycline H:Cdk7 (CAK). (23)[C-D3] Gadd45 et 14–3–3σ se fixent au complexe cycline B:Cdk1 et bloquent son activité kinase. (24)[A2] Chk1 est activée par les UV et les anomalies de réplication, alors que Chk2 est activée par les radiations ionisantes (IR). (25)[B1] Plk3 est activée par ATM et ATR et, comme Chk1 et Chk2, phosphoryle p53 sur la sérine 20 et Cdc25C sur la sérine 216. Plk3 peut être activée par la doxorubicine, les UV, et le stress oxydatif (H2O2). (26)[C4] Un sillon dans la région N-terminale de Mdm2 lie un segment peptidique dans la région N-terminale de p53, ce qui stimule la dégradation de p53 après ubiquitinylation (26a)[C4]. (27)[D4] ATM inactive l’interaction entre Mdm2 et p53 par phosphorylation de Mdm2. |
| Dans le texte | |
|
Figure 5. Inhibiteurs des Cdk et des points de surveillance du cycle cellulaire. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.