")
")
| Numéro |
Med Sci (Paris)
Volume 31, Numéro 4, Avril 2015
|
|
|---|---|---|
| Page(s) | 404 - 416 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20153104015 | |
| Publié en ligne | 8 mai 2015 | |
Potentiel thérapeutique de la neuromodulation optogénétique
Therapeutic potential of optogenetic neuromodulation
1
Équipe dynamique et pathophysiologie des réseaux neuronaux, centre interdisciplinaire de recherche en biologie, CNRS UMR 7241, Inserm U1050, Collège de France, 11, place Marcelin Berthelot, 75005 Paris, France
2
AP-HP, hôpital Henri Mondor, service de neurochirurgie, 94000 Créteil, France
3
Inserm, U955, équipe 14, 94000 Créteil, France
4
Université Paris Est, faculté de médecine, 94000 Créteil, France
5
CNRS UMR 8197, Inserm U1024, institut de biologie de l’école normale supérieure, 46, rue d’Ulm, 75005 Paris, France
a
marie.vdc@gmail.com
b
guillaume.dugue@ens.fr
Apparues dans le courant des années 2000, les techniques de neuromodulation optogénétique ont considérablement accru notre pouvoir d’analyse du fonctionnement des systèmes nerveux en permettant l’utilisation d’impulsions lumineuses pour l’excitation et l’inhibition de populations neuronales ciblées génétiquement. Après avoir été adoptées de manière fulgurante par la communauté des neurosciences fondamentales, ces techniques sont aujourd’hui en passe d’ouvrir de nouvelles perspectives thérapeutiques. La neuromodulation optogénétique est un outil de choix pour l’étude de la physiopathologie de maladies neurologiques et neuropsychiatriques dans un certain nombre de modèles animaux. Elle pourrait ainsi accélérer la découverte de nouvelles stratégies thérapeutiques. De nouveaux traitements employant des protocoles de neuromodulation optogénétique pourraient également voir le jour à moyen terme, dans le but de traiter des maladies telles que l’épilepsie pharmaco-résistante et certains types de dégénérescences rétiniennes héréditaires, pour lesquelles il n’existe pas de solution thérapeutique satisfaisante.
Abstract
Optogenetic neuromodulation techniques, which have emerged during the last 15 years, have considerably enhanced our ability to probe the functioning of neural circuits by allowing the excitation and inhibition of genetically-defined neuronal populations using light. Having gained tremendous popularity in the field of fundamental neuroscience, these techniques are now opening new therapeutic avenues. Optogenetic neuromodulation is a method of choice for studying the physiopathology of neurological and neuropsychiatric disorders in a range of animal models, and could accelerate the discovery of new therapeutic strategies. New therapeutic protocols employing optogenetic neuromodulation may also emerge in the near future, offering promising alternative approaches for disorders which lack appropriate treatments, such as pharmacoresistant epilepsy and inherited retinal degeneration.
Vignette (Photo © Nir Grossman, Imperial College London) http://www.imperial.ac.uk/people/nir.grossman06
Cet article est le second d’une série de deux. Le premier correspond à la référence [2] et a été publié dans le numéro de mars 2015 de m/s.
© 2015 médecine/sciences – Inserm
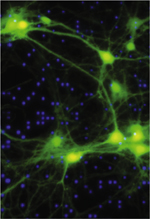
Principes et avantages de la neuromodulation optogénétique
L’optogénétique regroupe un ensemble d’approches utilisant la lumière pour observer ou interagir avec des structures ou fonctions biologiques ciblées génétiquement [1]. Dans le domaine des neurosciences expérimentales, cette technologie est principalement utilisée pour observer et contrôler (exciter ou inhiber) l’activité de populations neuronales spécifiques [2] (→).
(→) Voir la Synthèse de G.P. Dugué et L. Tricoire, m/s n°3, mars 2015, page 291
Le contrôle optogénétique de l’activité neuronale (neuromodulation optogénétique) implique que le potentiel transmembranaire des neurones cibles soit rendu photosensible par l’introduction d’un gène artificiel (Figure 1A). Ce gène code le plus souvent une protéine transmembranaire appartenant à la superfamille des opsines, dont l’association avec une molécule de rétinal (l’une des formes de la vitamine A) permet la formation d’une molécule photoréceptrice nommée rhodopsine. La rhodopsine la plus utilisée est la channelrhodopsine, un canal cationique sensible à la lumière bleue et provenant d’une famille d’algues unicellulaires. La photoactivation de ce canal par la lumière bleue produit l’apparition quasi instantanée d’un courant transmembranaire dépolarisant. Exprimée en quantité suffisante dans une cellule nerveuse, la channelrhodopsine permet de déclencher l’émission de potentiels d’action à l’aide d’impulsions lumineuses avec une précision temporelle de l’ordre de la milliseconde [2] (→).
(→) Voir la Synthèse de G.P. Dugué et L. Tricoire, m/s n°3, mars 2015, page 291
 |
Figure 1. Photosensibilisation de cellules nerveuses par transduction virale. A. Un transgène (ici codant la channelrhodopsine-2) est encapsidé dans des particules virales qui sont ensuite introduites dans une région cérébrale donnée par injection stéréotaxique. B. Pour des expériences de neuromodulation optogénétique sur des animaux en situation comportementale, un segment de fibre optique fixé dans un connecteur miniature peut être implanté sur la tête de l’animal. Lors de l’expérience, ce connecteur est relié à une source de lumière par l’intermédiaire d’un câble optique. C. La lumière délivrée par la fibre optique n’agit que sur les neurones photosensibilisés (ici en vert), c’est-à-dire ceux exprimant la protéine codée par le transgène. |
D’autres rhodopsines microbiennes qui jouent le rôle de pompes ioniques activées par la lumière verte ou jaune (halorhodopsines, archéorhodopsines et bactériorhodopsines) sont au contraire utilisées pour inhiber l’activité neuronale par hyperpolarisation de la membrane plasmique.
Les techniques actuelles de ciblage génétique permettent d’exprimer ces rhodopsines dans des catégories très spécifiques de neurones, qui deviennent ainsi sensibles à une longueur d’onde particulière (Figure 1C). La neuromodulation optogénétique permet d’atteindre des degrés de spécificité cellulaire et de précision spatiotemporelle inédits, bien supérieurs à la résolution des techniques utilisées jusque-là (pharmacologie, stimulations électriques ou magnétiques). Elle est ainsi devenue un outil incontournable pour l’analyse détaillée du fonctionnement des circuits neuronaux dans des modèles animaux aussi divers que le nématode, la drosophile, la souris et le primate non humain [3].
Le contrôle de l’activité de populations neuronales spécifiques au sein d’un parenchyme cérébral dense et hétérogène représente un enjeu méthodologique non seulement pour les neurosciences fondamentales, mais aussi pour l’étude et le traitement de certaines maladies du système nerveux. La neuromodulation optogénétique offre en effet la possibilité d’affiner la dissection des mécanismes physiopathologiques à l’œuvre dans les modèles animaux de certaines de ces maladies [4], dans le but d’identifier de nouvelles cibles thérapeutiques accessibles à des techniques de neuromodulation déjà validées en pratique clinique (pharmacologie, chirurgie lésionnelle, stimulations électriques en profondeur ou stimulation magnétique transcrânienne). La neuromodulation optogénétique pourrait également trouver sa place en tant que solution thérapeutique à part entière. Certaines maladies neurologiques ou neuropsychiatriques sont en effet clairement liées au dysfonctionnement de sous-circuits neuronaux particuliers, comme en témoignent un certain nombre d’études ayant employé des stimulations électriques profondes chez l’homme [5–8]. Cependant, les possibilités d’interventions ciblées à l’aide des techniques actuelles de neuromodulation sont fortement limitées. D’une part, les méthodes de stimulation par application d’un courant électrique ou d’un champ magnétique agissent de manière non sélective sur les différents types et compartiments neuronaux, et sont donc susceptibles de produire des effets indésirables par la stimulation de portions de circuits non pathologiques [9]. D’autre part, les méthodes de neuromodulation pharmacologique agissent à des échelles de temps largement supérieures à celles auxquelles opèrent les réseaux neuronaux (plusieurs minutes à plusieurs heures, contre quelques millisecondes). La neuromodulation optogénétique pourrait en principe se substituer à ces méthodes en offrant des voies d’intervention thérapeutiques hautement ciblées, rapides et réversibles.
Des obstacles à surmonter pour une utilisation de la neuromodulation optogénétique chez l’homme
La thérapie génique est habituellement définie comme l’introduction permanente d’un « gène médicament » chez un patient. Comme l’a souligné le Comité consultatif national d’éthique dans son avis numéro 36 du 22 juin 1993, cette modification ne doit en aucun cas modifier l’hérédité du malade. C’est la raison pour laquelle seule une thérapie génique dite « somatique » (n’affectant pas la lignée germinale) est envisageable chez l’homme. L’utilisation de la neuromodulation optogénétique nécessitera la validation d’un nouveau type de thérapie génique somatique pour au moins deux raisons : d’une part, le gène introduit ne sera pas à proprement parler un gène médicament, car le produit de son expression ne suffira pas, à lui seul, à fournir un effet thérapeutique (il faudra pour cela illuminer la portion de tissu génétiquement modifiée) ; d’autre part, le gène en question ne sera pas d’origine humaine, comme c’est le cas dans la plupart des essais cliniques actuels de thérapie génique, mais proviendra vraisemblablement d’une espèce microbienne (algue unicellulaire, bactérie ou archée). Afin d’introduire un ou plusieurs transgènes chez un patient, les essais récents de thérapie génique ont le plus souvent fait appel à des vecteurs viraux, dont l’efficacité demeure supérieure à celle des supports inertes (liposomes et polymères cationiques). Dans la pratique, l’utilisation de la neuromodulation optogénétique comme stratégie thérapeutique suppose de maîtriser trois types de risques : les risques inhérents à l’injection de vecteurs viraux (immunogénicité, possible recombinaison avec des virus sauvages, risques de diffusion dans l’entourage et potentiel effet délétère d’une insertion aléatoire dans le génome), les éventuels effets cytotoxiques de l’expression du transgène, ainsi que la phototoxicité de la longueur d’onde utilisée [10].
Les vecteurs viraux
Les vecteurs viraux utilisés en clinique sont essentiellement les adénovirus, les virus adéno-associés (AAV, pour adeno-associated virus) et certaines classes de rétrovirus (en particulier les lentivirus). Tous sont capables de s’insérer dans des cellules quiescentes (à l’exception des rétrovirus non lentiviraux) et peuvent donc être utilisés pour transduire des cellules nerveuses différenciées. Les adénovirus permettent de délivrer des transgènes de grande taille (jusqu’à 30 kb), mais leur utilisation est grandement limitée par les réactions inflammatoires et immunitaires induites par l’injection de particules adénovirales. Cette forte immunogénicité s’explique notamment par le fait qu’une grande partie de la population (environ 80 %) possède des anticorps spécifiques issus d’une infection préalable par des souches sauvages d’adénovirus. Elle serait responsable de la perte rapide d’expression du transgène et de la faible efficacité des ré-administrations [11]. Les adénovirus ont également le désavantage de présenter un tropisme pour les cellules gliales, ce qui limite leur utilité dans le cadre de manipulations visant des populations neuronales [12]. Les AAV induisent une réponse immunitaire beaucoup plus faible que les adénovirus et ont l’avantage de présenter un tropisme neuronal satisfaisant. Leur petite taille (20 nm) leur permet de diffuser plus aisément que les autres vecteurs viraux et ainsi de transduire des volumes plus étendus de tissu nerveux. Plusieurs essais thérapeutiques de phase I ont permis d’établir l’innocuité de l’injection d’AAV dans le système nerveux central [13–15]. Le principal désavantage des AAV réside dans leur faible capacité de transport (jusqu’à 5 kb). Contrairement aux adénovirus et aux AAV, le transgène véhiculé par un lentivirus s’insère de manière stable dans le génome. Toutefois ce processus d’intégration, souvent aléatoire, peut en théorie engendrer des mutations insertionnelles, ce qui explique pourquoi les lentivirus ont jusqu’à présent été délaissés dans la plupart des essais cliniques [16]. Comparés aux AAV, les lentivirus présentent pourtant l’avantage d’assurer le transfert d’une plus grande quantité d’ADN (jusqu’à 8 kb). Ils induisent également une réaction immunitaire minime qui les rend compatibles avec des injections répétées [17]. Enfin, compte tenu de leur diffusion plus réduite que celle des AAV, ils sont adaptés au ciblage de petits volumes cérébraux. En 2014, un essai thérapeutique de phase I/II pour une thérapie génique dopaminergique dans la maladie de Parkinson a établi pour la première fois l’innocuité d’un lentivirus, l’EIAV (equine infectious anemia virus), directement administré dans le système nerveux central [18].
Risques associés à la surexpression des rhodopsines
Les rhodopsines microbiennes utilisées dans la plupart des protocoles de neuromodulation optogénétique produisent des courants unitaires de petite taille. L’efficacité de ces approches réside donc en grande partie dans l’utilisation de méthodes de transgenèse pouvant assurer de forts niveaux d’expression, telles que la transduction virale. Cependant, les effets d’une surexpression de ce type de protéine sont encore largement inexplorés à l’échelle cellulaire [10]. On peut ainsi imaginer que les propriétés d’un neurone soient modifiées au-delà d’un seuil d’expression critique, par accumulation de la rhodopsine dans des compartiments intracellulaires, ou par modification des propriétés de la membrane plasmique. Une étude chez le rongeur a montré qu’une surexpression massive, prolongée et intentionnelle de la channelrhodopsine-2 (ChR2) de l’algue Chlamydomonas reinhardtii, induite dès le stade embryonnaire par électroporation in utero, pouvait entraîner des anomalies dans la morphologie de neurones corticaux au stade adulte [19]. En revanche, des études employant un lentivirus pour l’expression de la ChR2 chez le primate non humain se sont montrées rassurantes plus d’un an après l’injection du vecteur viral en termes de réaction inflammatoire, et de morphologie et densité des neurones transduits [20]. Notons que des effets potentiellement délétères pourraient également survenir non pas par surexpression des rhodopsines, mais par un influx excessif d’un type d’ion particulier au cours de stimulations lumineuses chroniques. On peut se demander, par exemple, si l’activation répétée de la ChR2, dont on connaît la perméabilité non négligeable au calcium, pourrait être susceptible de recruter des voies de signalisation pro-apoptotiques dépendantes du calcium.
Risques associés à l’illumination du tissu nerveux
L’illumination de zones profondes du cerveau (au-delà de plusieurs millimètres sous la surface) ne peut se faire qu’au moyen de guides de lumière tels que des fibres optiques. Le diamètre de ces fibres ne mesure typiquement que quelques centaines de micromètres (200-400 µm) afin de minimiser les dommages provoqués par leur insertion dans le tissu nerveux. Pour illuminer des volumes suffisamment étendus, il est nécessaire de concentrer une intensité lumineuse relativement forte dans ces fibres optiques (l’irradiance en sortie de fibre est typiquement de plusieurs centaines de milliwatts par millimètre carré). Le fait de concentrer ainsi la lumière expose au danger d’induire un échauffement local du parenchyme cérébral à proximité de l’extrémité de la fibre optique [10]. Ainsi, pour la lumière bleue, il n’est pas rare d’observer une modulation artéfactuelle de l’activité neuronale (en l’absence de photosensibilisation préalable) au-delà d’un certain seuil d’irradiance (300-500 mW/mm2). La quantité de lumière nécessaire est d’autant plus faible que le niveau d’expression de la rhodopsine est élevé, de sorte que les véritables conditions optimales pour une expérience de neuromodulation optogénétique résultent d’un compromis entre niveau d’expression du transgène, intensité lumineuse et volume photomodulé. Le jeu idéal de paramètres doit ainsi être choisi de manière à maximiser l’effet désiré (la modulation d’une population particulière de neurones) tout en minimisant les risques liés à l’expression du transgène et à l’illumination du tissu.
Malgré ces contraintes, il existe de bonnes raisons de penser que la neuromodulation optogénétique offre des perspectives thérapeutiques crédibles. Les outils neurochirurgicaux pour pratiquer des injections stéréotaxiques intracrâniennes chez l’homme existent déjà (Figure 2A). Ils pourront permettre la transduction de cellules nerveuses dans des régions cérébrales précisément délimitées, à l’aide de vecteurs viraux ayant déjà démontré leur innocuité chez le patient [21]. Les éventuels problèmes de phototoxicité pourraient être grandement atténués par l’utilisation de rhodopsines dont le spectre d’activation est décalé vers le rouge (permettant d’utiliser un rayonnement lumineux plus pénétrant et moins porteur d’énergie) [22–24], et/ou présentant une sensibilité à la lumière accrue [25, 26]. La mise au point de dispositifs d’illumination du tissu nerveux, qu’il s’agisse de fibres optiques implantées de manière prolongée ou de tapis de diodes électroluminescentes déposés sur la dure-mère, pourra bénéficier de l’expérience acquise avec des dispositifs déjà validés en clinique pour la stimulation électrique en profondeur et l’électrocorticographie (Figure 2B). Enfin, l’efficacité de la neuromodulation optogénétique a déjà été évaluée avec succès chez le primate non humain, et ce de plusieurs manières : par électrophysiologie [27–29], par imagerie par résonance magnétique fonctionnelle [21] et par des études comportementales [21, 30]. Le bénéfice thérapeutique de protocoles utilisant la neuromodulation optogénétique n’a en revanche été évalué que dans un petit nombre de modèles de maladies neurologiques chez le rongeur.
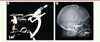 |
Figure 2. Exemples de dispositifs cliniques utilisés actuellement pour les injections et stimulations intracrâniennes. A. Dispositif d’injection stéréotaxique de vecteurs viraux. B. Électrodes de stimulation implantées dans le noyau sous-thalamique d’un patient parkinsonien et révélées par radiographie. |
Approches optogénétiques dans la maladie de Parkinson
La maladie de Parkinson est une maladie neurodégénérative qui touche près de 150 000 personnes en France (plus de six millions de personnes dans le monde). Elle est causée par la perte progressive des neurones dopaminergiques d’un noyau mésencéphalique appelé « substance noire ». Ces neurones projettent vers un ensemble de structures sous-corticales regroupées sous l’appellation « ganglions de la base » (Figure 3), impliquées dans des fonctions cognitivo-motrices telles que le contrôle et l’exécution de mouvements volontaires, la sélection de programmes moteurs, émotionnels et cognitifs adaptés au contexte environnemental, l’apprentissage procédural et la formation d’habitudes comportementales. La dénervation dopaminergique des ganglions de la base entraîne des modifications pathologiques de leur activité à l’origine d’une triade de symptômes moteurs gravement handicapants (akinésie, rigidité et tremblements), ainsi que d’une série de désordres cognitifs pouvant aller de symptômes légers (troubles de l’attention, de la mémoire ou de fonctions exécutives telles que la planification des actions du sujet) jusqu’à la démence [31, 32].
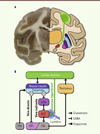 |
Figure 3. Implication du faisceau cortico-sous-thalamique dans le mécanisme d’action de la stimulation du noyau sous-thalamique chez le patient parkinsonien. A. Photographie (à gauche) et schéma (à droite) représentant une coupe coronale de cerveau humain. Une partie des ganglions de la base sont représentés en couleur sur le schéma. Le code couleur des différents noyaux est identique à celui du diagramme en B. Le faisceau cortico-sous-thalamique (voie hyperdirecte) est représenté en vert. B. Diagramme de connectivité simplifié des ganglions de la base représentant la voie directe, la voie indirecte, ainsi que la voie hyperdirecte. Chez la souris, la photostimulation (flèche bleue) sélective des afférences de la voie hyperdirecte dans le noyau sous-thalamique suffit à reproduire les effets antiparkinsoniens de la stimulation électrique du noyau sous-thalamique [37]. D1 et D2 : neurones striataux exprimant les récepteurs dopaminergiques D1 et D2 respectivement ; GPe : globus pallidus externe ; GPi : globus pallidus interne ; NST : noyau sous-thalamique ; SNc : substance noire pars compacta ; SNr : substance noire pars reticulata (adapté de [84]). |
La principale cible des neurones dopaminergiques de la substance noire est le striatum (noyau caudé et putamen chez l’homme), point d’entrée des afférences corticales dans les circuits des ganglions de la base. Le striatum contient deux populations de neurones de projection exprimant des récepteurs dopaminergiques de sous-types différents (D1 et D2). Selon le modèle classique, les neurones exprimant le récepteur D1 sont responsables de l’activation d’une voie cortico-nigrale dite directe, dont l’effet est de faciliter l’exécution des mouvements (effet prokinétique), et sont activés par la dopamine. Les neurones exprimant le récepteur D2 activent une voie cortico-pallido-nigrale indirecte, qui tend à inhiber le mouvement (effet anti-kinétique), et sont inhibés par la dopamine (Figure 3B). Ainsi, la dénervation dopaminergique diminuerait l’activité de la voie directe prokinétique, et activerait, par désinhibition, l’activité de la voie indirecte antikinétique, l’ensemble concourant à l’inhibition des mouvements. Ce modèle est régulièrement remis en cause par des données contredisant la ségrégation anatomique des voies directe et indirecte. La modulation optogénétique a permis de dépasser ce débat en testant directement chez la souris les effets de l’activation des neurones striataux exprimant les récepteurs D1 ou D2 [33]. Ce type de manipulation est inenvisageable par le biais de stimulations électriques, car les deux populations de neurones striataux sont mêlées dans la plus grande partie du striatum. Les résultats, tant sur l’activité locomotrice des animaux que sur l’activité des structures de sortie des ganglions de la base (où les voies directe et indirecte se rejoignent), ont permis d’établir la validité fonctionnelle du modèle « classique », indépendamment des controverses anatomiques.
À l’heure actuelle, la principale approche thérapeutique pour lutter contre le déficit en dopamine dans la maladie de Parkinson consiste à administrer son précurseur naturel direct, la lévodopa (L-DOPA), par voie orale. La L-DOPA est capable de franchir la barrière hémato-céphalique et peut ainsi être métabolisée en dopamine et sécrétée par les terminaisons des neurones dopaminergiques résiduels. Ce traitement parvient à corriger l’activité pathologique des ganglions de la base et à alléger les symptômes moteurs, mais il devient inefficace à moyen terme en entraînant des fluctuations motrices ainsi que des mouvements anormaux involontaires appelés dyskinésies, tout aussi invalidants que la maladie elle-même [34]. C’est à ce stade que l’on propose à certains patients (seuls 15 % sont éligibles) un traitement neurochirurgical alternatif mis au point dans les années 1990 : la stimulation cérébrale profonde (SCP) électrique à haute fréquence de l’un des constituants des ganglions de la base, le noyau sous-thalamique (NST) [35]. Bien que les effets thérapeutiques de la SCP sur les symptômes moteurs se soient révélés spectaculaires dès les premiers essais cliniques, les mécanismes par lesquels ce protocole de stimulation normalise les activités pathologiques n’ont été élucidés que partiellement [36].
L’utilisation de la neuromodulation optogénétique dans un modèle murin de la maladie de Parkinson a permis récemment d’identifier la voie neuronale dont la stimulation est vraisemblablement responsable de l’effet thérapeutique de la SCP du NST [37]. En utilisant une lignée de souris transgéniques dans laquelle la ChR2 est exprimée dans les neurones de projection du néocortex, mais pas dans les neurones du NST, des chercheurs de l’université Stanford ont pu montrer que la photostimulation du faisceau moteur cortico-sous-thalamique (appelé faisceau « hyperdirect » ; voir Figure 3 ) suffit à reproduire les effets de la SCP du NST. Ce résultat permet d’établir un lien crucial entre les effets de la SCP et ceux observés au moyen de stimulations corticales électriques chez le primate non humain [38], ou lors d’essais de stimulation magnétique transcrânienne du cortex moteur chez des patients parkinsoniens [39]. La stimulation optogénétique pourrait offrir une alternative thérapeutique intéressante aux stimulations électriques, car elle permettrait de ne cibler que la composante motrice du faisceau cortico-sous-thalamique (celle émanant du cortex moteur), contrairement à la SCP du NST, qui affecte potentiellement les composantes associatives et limbiques des afférences corticales dans le NST, pouvant engendrer des dépressions, hypomanies, et syndromes confusionnels [9]. Il est important de noter qu’une stratégie de stimulation optogénétique qui nécessiterait l’implantation de fibres optiques afin de guider la lumière jusqu’au NST se heurterait aux mêmes complications que celles qui sont associées dans certains cas à l’implantation d’électrodes pour la SCP (trajectoire désaxée de l’électrode, hématomes intracérébraux et infections [40]). C’est pourquoi la possibilité de ne cibler l’expression de la ChR2 que dans les neurones corticaux à l’origine du faisceau hyperdirect (par exemple par infection rétrograde depuis le NST [20]) permettrait de traiter les patients par simple illumination du cortex moteur, à l’aide d’outils beaucoup moins invasifs tels que des tapis de diodes électroluminescentes disposés en position épidurale.
Approches optogénétiques dans le traitement de l’épilepsie
Options thérapeutiques actuellement disponibles pour le traitement de l'épilepsie
L’épilepsie est une maladie neurologique chronique affectant environ 1 % de la population mondiale. Elle se caractérise par l’apparition intermittente de crises, ou « ictus épileptiques », au cours desquelles l’activité de portions plus ou moins larges de tissu nerveux devient anormalement intense (paroxystique) et/ou hypersynchrone. De nombreux agents pharmacologiques visant à diminuer l’excitabilité des réseaux neuronaux sont utilisés dans le traitement de l’épilepsie. Ces traitements antiépileptiques présentent toutefois au moins deux inconvénients majeurs. D’une part, leur effet prolongé ne permet pas de limiter leur action à la durée d’une crise, de sorte que la prise d’antiépileptiques affecte le fonctionnement normal du cerveau entre les ictus, donnant lieu à des effets secondaires indésirables (vertiges, somnolence, troubles de la vision). D’autre part, ces médicaments ont une action limitée face à la diversité des types cliniques d’épilepsie, dont on considère classiquement qu’au moins 20 à 30 % sont - ou deviennent - réfractaires au traitement (on parle d’épilepsie pharmaco-résistante) [41]. La neurochirurgie résective constitue actuellement la principale option alternative pour les patients pharmaco-résistants. Elle consiste à pratiquer soit l’ablation du foyer épileptique (cortectomie) ou de la lésion responsable de l’épilepsie (lésionectomie), soit à sectionner les voies de propagation préférentielles de l’activité épileptique (comme par exemple dans le cas de la callosotomie, qui vise à empêcher la bilatéralisation de la décharge épileptique par le corps calleux). Ce type de traitement revêt un caractère définitif, se traduisant au mieux par la diminution ou l’arrêt des crises, mais pouvant également s’accompagner d’un certain nombre de déficits post-opératoires persistants.
L’utilisation de la SCP pour le traitement de l’épilepsie pharmaco-résistante a fait l’objet de plusieurs dizaines d’études chez l’homme au cours des dix dernières années [42], mais seule l’efficacité de la stimulation du noyau antérieur du thalamus pour le traitement des épilepsies focales a véritablement été démontrée jusqu’à présent [43]. Bien que les mécanismes d’action de la SCP soient encore mal connus, ce type de traitement est susceptible de diminuer la sévérité et la fréquence des crises de plusieurs manières : en normalisant directement l’activité du foyer épileptique, en agissant au niveau des voies et nœuds de propagation de l’activité épileptique, ou enfin en activant des mécanismes de plasticité ayant pour effet de diminuer l’excitabilité des circuits épileptogènes. Les stimulations électriques peuvent être administrées sous la forme de protocoles continus, mais pourront très prochainement être déclenchées par un système d’enregistrement électrophysiologique chargé de détecter l’arrivée des crises (on parle de système en « boucle fermée »). Les systèmes en boucle fermée représentent un réel progrès puisqu’ils permettent de ne pas affecter l’activité du cerveau entre les ictus, et idéalement de stopper les crises avant l’expression de symptômes moteurs dangereux pour le patient (tels que des convulsions ou une perte de conscience). Un premier système de stimulation en boucle fermée pour le traitement de l’épilepsie a récemment reçu l’approbation de la Food and drug administration pour une mise sur le marché aux États-Unis.
Perspectives thérapeutiques offertes par les outils optogénétiques
Comme dans le cas du traitement de la maladie de Parkinson par la SCP à haute fréquence, la neuromodulation optogénétique pourrait se substituer avantageusement à la SCP dans le traitement de l’épilepsie pharmaco-résistante. En permettant de contrôler sélectivement neurones excitateurs ou inhibiteurs, les techniques de stimulation ou d’inhibition optogénétiques seraient en effet plus à même de corriger le déséquilibre de la balance excitation/inhibition caractéristique de l’état épileptique.
Une première stratégie consiste à utiliser des outils optogénétiques hyperpolarisants pour inhiber directement les cellules excitatrices dont l’hyperactivité est responsable de l’apparition et de la propagation de l’activité ictale (Figure 4A). Dès 2009, une première étude a démontré que l’expression ciblée de l’halorhodopsine de l’archée Natronomonas pharaonis (NpHR) pouvait être utilisée pour réduire ou inhiber des bouffées d’activité épileptiforme induites par stimulation électrique dans des cultures organotypiques d’hippocampe [44]. Cette approche a par la suite été validée in vivo dans différents modèles murins, obtenus par injection de substances épileptogènes. Dans ces expériences, différentes versions de la NpHR ont été utilisées pour inhiber de manière prolongée l’activité des neurones excitateurs dans le foyer épileptique (cortical ou hippocampique), aboutissant à une réduction de 20 à 50 % de la fréquence des bouffées épileptiformes [45, 46], ou une augmentation de la latence de leur apparition de l’ordre de 30 % [47] durant les phases d’illumination. Une deuxième stratégie consiste à contrecarrer l’hyperactivité des neurones excitateurs durant les crises d’épilepsie en recrutant le plus largement possible les réseaux d’interneurones inhibiteurs locaux (Figure 4B). Une étude récente a montré que dans des tranches de tissu hippocampique, la fréquence moyenne des bouffées d’activité épileptiforme induites par un traitement pharmacologique est réduite de moitié pendant des phases de photostimulation de 10 secondes des interneurones inhibiteurs [48]. Le mécanisme par lequel la stimulation de ces neurones conduit à une normalisation de l’activité du tissu n’a cependant pas été clairement établi. En effet les auteurs suggèrent que le neurotransmetteur relargué par ces interneurones, le GABA, a un effet excitateur plutôt qu’inhibiteur au sein du tissu épileptique (suite à un déséquilibre dans la répartition des ions chlorure de part et d’autre de la membrane plasmique), mais cette question reste débattue [49, 50].
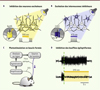 |
Figure 4. Stratégies optogénétiques pour le traitement de l’épilepsie. A-B. Deux options sont possibles : inhiber les neurones excitateurs impliqués dans la genèse ou la propagation de l’activité ictale (A) , ou exciter les interneurones inhibiteurs locaux (B) , par expression spécifique de pompes hyperpolarisantes (telles que la NpHR) ou d’outils dépolarisants (tels que la ChR2) respectivement. C. Principe de la neuromodulation optogénétique en boucle fermée, qui permet de n’illuminer le tissu nerveux que lorsqu’une crise est détectée. EEG : électro-encéphalogramme. D. Exemples de tracés électrophysiologiques démontrant l’efficacité de l’inhibition optogénétique en boucle fermée dans un modèle d’épilepsie du lobe temporal chez la souris. La ligne verte pointillée indique le moment où l’algorithme détecte l’arrivée d’une crise. L’activité épileptiforme peut être stoppée lorsque cet algorithme déclenche l’illumination du tissu. Reproduit d’après [52]. |
Les résultats les plus encourageants ont été obtenus récemment chez des animaux vigiles à l’aide de systèmes de neuromodulation optogénétique en boucle fermée. Ces systèmes fonctionnent en réalisant une analyse spectrale en temps réel du signal électroencéphalographique intracrânien, et ne déclenchent le protocole d’illumination que lorsqu’un épisode d’activité ictale est détecté (Figure 4C–D). Dans une première étude, le laboratoire du Dr John Huguenard à l’université Stanford s’est intéressé au lien fonctionnel entre ischémie corticale focale (provoquée classiquement par un accident vasculaire cérébral) et apparition de crises épileptiques (l’épilepsie chronique étant l’une des complications connues de l’accident vasculaire cérébral ischémique, en particulier chez les patients âgés). Le but de cette étude était d’évaluer le rôle des connexions réciproques cortico-thalamiques dans la propagation de l’activité ictale, et de tester l’efficacité d’une inactivation optogénétique du thalamus pour le traitement de ce type d’épilepsie. Dans cette étude, une ischémie focale permanente était obtenue chez des rats par photothrombose corticale. Plusieurs semaines plus tard, ces animaux développaient une épilepsie chronique caractérisée par l’apparition de crises spontanées. Une large fraction des neurones thalamocorticaux (excitateurs) interconnectés avec la région corticale lésée étaient ensuite photosensibilisés par expression de la NpHR à l’aide d’un vecteur viral. Le protocole d’illumination de ces neurones en boucle fermée s’est révélé capable de stopper les crises d’épilepsie moins d’une seconde après leur apparition, et ce jusqu’à un an après implantation du système chez les animaux [51]. Dans une deuxième étude, un modèle d’épilepsie du lobe temporal était obtenu chez la souris par lésion pharmacologique focale de l’hippocampe à l’acide kaïnique. Les neurones de l’hippocampe étaient ensuite photosensibilisés de manière sélective, par expression soit de la NpHR dans les neurones excitateurs, soit de la ChR2 dans une catégorie spécifique d’interneurones inhibiteurs. L’illumination de ces neurones au tout début des épisodes d’activité ictale a permis de stopper plus de la moitié des crises dans les deux cas (en une seconde seulement par inhibition des neurones excitateurs exprimant la NpHR et en cinq secondes par excitation des interneurones inhibiteurs exprimant la ChR2) [52].
Traitement optogénétique des rétinopathies pigmentaires
Les rétinopathies pigmentaires forment un ensemble hétérogène de maladies héréditaires de la rétine caractérisées par la dégénérescence progressive des cellules photoréceptrices situées au contact de l’épithélium pigmentaire (Figure 5B) [53]. Ce type de maladie touche environ une personne sur 4 000 dans le monde et peut être causé par la mutation de plus de 50 gènes différents [54, 55]. Les premiers symptômes visuels apparaissent classiquement entre 10 et 30 ans avec la perte des photorécepteurs à bâtonnets (responsables de la vision en très faible luminance, appelée vision scotopique). Ils s’aggravent ensuite progressivement avec la dégénérescence des photorécepteurs à cônes (responsables de la vision diurne, appelée vision photopique), jusqu’à aboutir le plus souvent à une cécité bilatérale complète.
 |
Figure 5. Différentes options pour le traitement optogénétique des rétinopathies pigmentaires. A. Schéma anatomique de l’œil humain. Les vecteurs viraux peuvent être injectés dans l’humeur vitrée (injection intra-vitréenne) ou sous la rétine. Les injections sous-rétiniennes présentent l’inconvénient d’endommager mécaniquement un tissu déjà fragilisé par le processus de dégénérescence, et de ne pouvoir atteindre que des surfaces restreintes de rétine. B. Schéma simplifié du circuit rétinien, montrant les deux principaux types de circuits (ON et OFF) en aval des photorécepteurs à cônes. Plusieurs types neuronaux ne sont pas représentés (cellules amacrines et cellules horizontales). C. Stade avancé de dégénérescence rétinienne dans lequel seuls certains photorécepteurs à cônes subsistent sous la forme de cellules atrophiées (cônes dormants). La rephotosensibilisation du circuit peut s’appuyer sur l’expression de la ChR2 dans les cellules ganglionnaires (accessibles par voie intra-vitréenne à l’aide d’AAV uniquement), par expression de la ChR2 dans les cellules bipolaires de type ON ou de NpHR dans les cônes dormants (types cellulaires accessibles par injection sous-rétinienne de plusieurs classes de virus, et par injection intra-vitréenne d’AAV présentant un fort pouvoir de diffusion). |
Stratégies thérapeutiques pour le traitement des rétinopathies pigmentaires
La recherche de stratégies thérapeutiques pour ce type de rétinopathie s’est orientée vers différentes possibilités. La stimulation électrique des cellules ganglionnaires à l’aide d’une « prothèse rétinienne » est une première possibilité. Ces cellules, situées en aval du circuit rétinien, donnent naissance au nerf optique (Figure 5B) et sont épargnées par le processus de dégénérescence. Les prothèses rétiniennes sont constituées d’une couche de microélectrodes implantées à proximité de la fovéa et reliées, soit directement à une couche de microphotodiodes, soit à une caméra vidéo via un module chargé d’analyser l’image et de définir les patrons de stimulation en temps réel [56]. Ce concept a donné des résultats encourageants lors d’essais cliniques qui ont abouti à la mise sur le marché de la prothèse Argus II. Mais l’acuité visuelle obtenue à l’aide de ce type d’appareil reste en deçà du seuil de cécité défini par l’Organisation mondiale de la santé [57]. Une deuxième possibilité est l’utilisation d’une thérapie cellulaire consistant à transplanter des cellules pouvant se différencier en cellules photoréceptrices (cellules souches ou cellules progénitrices de la rétine) et remplacer les cellules endommagées [58]. Un autre type de thérapie cellulaire consiste à combattre le processus de dégénérescence grâce à la greffe sous-rétinienne de cellules pouvant libérer des facteurs de survie [59]. Enfin une dernière possibilité consiste à concevoir des stratégies de thérapie génique à caractère correctif (visant à insérer dans les cellules atteintes une version saine du ou des gènes défectueux), ou simplement à visée symptomatique (neuroprotection par expression de facteurs améliorant la survie des photorécepteurs) [60–63]. Cet axe de recherche a été exploré de manière intense, notamment parce que l’œil est un organe facile d’accès et qui semble bien tolérer les infections par les AAV [64]. Le protocole de thérapie génique corrective le plus prometteur à l’heure actuelle est celui visant à lutter contre l’amaurose congénitale de Leber par remplacement du gène RPE65. Ce type d’approche ne fonctionne cependant que pour des mutations récessives (dans le cas d’une mutation dominante, la présence de l’allèle muté suffit à tuer la cellule). La thérapie génique corrective se heurte également à la diversité des gènes dont la mutation peut être à l’origine de la maladie (le gène défectueux doit être identifié au cas par cas, et sa version saine doit être suffisamment petite pour pouvoir être encapsidée dans un vecteur viral).
La neuromodulation optogénétique : une option thérapeutique prometteuse ?
L’essor de la neuromodulation optogénétique a permis l’émergence d’une nouvelle voie thérapeutique visant à restaurer la photosensibilité du circuit rétinien altéré. Les premières études ont cherché à évaluer le bénéfice d’une photosensibilisation des cellules ganglionnaires, dont seule une petite minorité exprimant la mélanopsine est naturellement photosensible (ces cellules sont impliquées dans des fonctions non visuelles telles que le réflexe pupillaire et la régulation du rythme circadien). Bien que l’expression de la ChR2 dans les cellules ganglionnaires de souris rd1 (le modèle murin de dégénérescence rétinienne le plus utilisé) ait permis de restaurer des réponses électrophysiologiques en réponse à des flashs de lumière bleue jusque dans le cortex visuel [65, 66], les résultats disponibles à ce jour n’ont pas révélé d’amélioration significative dans des tâches comportementales de discrimination visuelle [66, 67]. Dans une autre série d’expériences en revanche, l’expression ectopique de la mélanopsine dans une large fraction des cellules ganglionnaires de souris rd1 a permis de restaurer une sensibilité comportementale à des différences de luminance [68]. Ce résultat est probablement dû au fait que la mélanopsine confère aux cellules ganglionnaires une photosensibilité très supérieure à celle résultant de l’expression de la ChR2. À la différence de la ChR2, une seule molécule de mélanopsine est en effet capable de moduler de nombreuses conductances membranaires par l’intermédiaire d’une cascade de signalisation intracellulaire faisant office de système amplificateur. Les réponses électrophysiologiques induites par photoactivation de la mélanopsine sont cependant trop lentes pour pouvoir espérer la restauration de fonctions visuelles fines. Plus récemment, l’expression d’un outil photochimique (le canal contrôlé par la lumière LiGluR) dans les cellules ganglionnaires a également donné des résultats positifs dans un test d’évitement d’une source de lumière [69]. Cette approche nécessite cependant l’injection d’un ligand photochromique, chaque injection permettant d’obtenir un effet comportemental pendant seulement deux jours.
Malgré ces résultats encourageants, la photosensibilisation indiscriminée des cellules ganglionnaires comporte un inconvénient de taille. Cette population de cellules contient en effet trois sous-populations fonctionnellement distinctes, l’une déchargeant en réponse à des incréments de lumière (cellules « ON »), l’autre en réponse à des décréments de lumière (cellules « OFF ») et la dernière réagissant aux deux situations (cellules « ON-OFF »). Dans les stratégies présentées ci-dessus, l’ensemble des cellules ganglionnaires sont reprogrammées en cellules « ON », et le message transmis au reste du cerveau diffère donc fortement de celui transmis par une information rétinienne normale. Plus généralement, il est important de noter que la rétine ne transmet pas l’information brute issue des photorécepteurs, mais en extrait des éléments saillants (forme, couleur, mouvement) grâce à des capacités de traitement élaborées. Afin de préserver au maximum la qualité du signal transmis au cerveau, il est donc souhaitable d’intervenir le plus en amont possible dans le circuit. Le groupe du Dr Botond Roska a montré qu’il était possible de photosensibiliser sélectivement les cellules bipolaires de type ON, situées entre les cônes et les cellules ganglionnaires de type ON (Figure 5C), par électroporation d’un plasmide contenant la séquence de la ChR2 sous le contrôle du promoteur d’un gène spécifiquement exprimé par ces cellules [70]. Bien que l’efficacité de cette stratégie se soit révélée faible (seules 7 % des cellules cibles exprimaient la ChR2 dans les zones électroporées), les souris rd1 traitées ont cependant recouvré la capacité de suivre le mouvement de rayures lumineuses verticales par des mouvements de la tête (réponse optomotrice). Des études ultérieures ont montré qu’il était possible d’exprimer la ChR2 plus efficacement dans les cellules bipolaires de type ON à l’aide d’AAV dont la capside a été modifiée afin d’améliorer leur diffusion au sein du tissu rétinien [71–73]. Deux d’entre elles ont validé cette stratégie à l’aide d’enregistrements in vivo et/ou de tests comportementaux. Dans la première étude, conduite par le groupe du Dr Alan Horsager, des injections sous-rétiniennes d’AAV ont permis de restaurer des réponses ON dans les cellules ganglionnaires ainsi que des scores proches de ceux de souris saines dans un test d’orientation guidé par une source lumineuse (pour des fortes intensités lumineuses) [73]. Dans la deuxième étude, le groupe du Dr Deniz Dalkara (voir le Dossier technique qui sera publié le prochain numéro de m/s [n° 5, mai 2015] [85]) est parvenu à transduire spécifiquement plus de la moitié des cellules bipolaires de type ON par injection intra-vitréenne (une voie d’administration moins traumatique que l’injection sous-rétinienne, qui tend à détacher les photorécepteurs de l’épithélium pigmentaire sous-jacent ; Figure 5A ) [72]. Cette procédure a permis de restaurer non seulement des réponses de type ON dans les cellules ganglionnaires et les neurones du cortex visuel des souris rd1 traitées, mais aussi des réponses de type OFF (il est connu que les circuits ON et OFF fonctionnent en interaction). Elle a également permis aux animaux traités de recouvrer un comportement d’évitement face à une source de lumière intense.
Il est envisageable d’intervenir encore plus en amont, au niveau des cônes résiduels observés dans de nombreux cas de rétinopathie pigmentaire. Ces cônes « dormants » subsistent dans la région fovéale jusqu’à des stades avancés de la maladie, sous la forme de corps cellulaires ayant perdu leur segment photosensible (Figure 5C). L’équipe du Dr Botond Roska a démontré qu’il était possible de réactiver ces cônes dormants par injection sous-rétinienne d’un AAV codant la NpHR sous le contrôle d’un promoteur spécifique des cônes (l’utilisation de la NpHR permet de reproduire l’hyperpolarisation induite normalement dans les cônes par la lumière ; la NpHR doit être exprimée de manière très spécifique afin de ne pas inhiber le transfert de l’information dans le reste du réseau) [74]. Des enregistrements électrophysiologiques ont montré que cette technique permet de restaurer les patrons de décharge naturels des cellules ganglionnaires en réponse à des stimulus visuels, avec toutefois une sensibilité très significativement inférieure (trois ordres de grandeur) à celle d’une rétine saine. Des tests comportementaux ont révélé que les souris traitées étaient capables de détecter des changements de luminosité et pouvaient recouvrer une réponse optomotrice.
Des obstacles à surmonter pour une rephotosensibilisation efficace de la rétine par des outils optogénétiques
L’ensemble des études réalisées ces huit dernières années montrent que la neuromodulation optogénétique est une option thérapeutique prometteuse pour le traitement d’un certain nombre de dégénérescences rétiniennes. Des obstacles restent cependant à surmonter avant de pouvoir envisager des traitements offrant un réel bénéfice thérapeutique chez l’homme. Les outils optogénétiques disponibles à l’heure actuelle offrent des seuils de photosensibilité très nettement inférieurs à ceux d’une rétine saine, ainsi qu’une gamme de sensibilité spectrale réduite. Ces inconvénients orientent une partie des efforts actuels de bio-ingénierie des rhodopsines microbiennes, dans le but de maximiser leurs photocourants et de diversifier leurs propriétés spectrales. Ils soulignent également l’importance d’obtenir des forts niveaux d’expression et donc d’optimiser les techniques de transduction virale des cellules rétiniennes. À l’heure actuelle, les bénéfices tirés de l’utilisation d’AAV dans des modèles murins (en particulier la possibilité d’obtenir de bons niveaux d’expression par injection intra-vitréenne) sont difficiles à reproduire chez le primate [63]. Notons enfin que les outils optogénétiques ne permettent pas de reproduire les fantastiques capacités d’adaptation des photorécepteurs à une très large gamme d’intensités lumineuses (assurées par des mécanismes moléculaires de régulation des cascades de phototransduction en aval des rhodopsines) qui permettent d’adapter la vision aux fortes variations de luminance rencontrées en conditions naturelles.
L’un des principaux défis à surmonter réside dans la complexité du circuit rétinien. Afin d’intervenir de manière réellement spécifique, il faudra affiner et peut-être combiner les stratégies permettant de cibler des sous-populations neuronales fonctionnellement distinctes (par exemple pour exprimer des outils dépolarisants et hyperpolarisants dans les cellules ON et OFF respectivement). Afin de pallier la faible sensibilité des rhodopsines microbiennes et de prendre en compte la complexité du traitement de l’information visuelle par la rétine, certains laboratoires envisagent l’utilisation de systèmes portatifs chargés de convertir un flux vidéo en une série de stimulations lumineuses de forte intensité destinées à recréer un patron de décharge physiologique dans le sous-circuit photosensibilisé. L’holographie digitale ou des matrices de micro-miroirs par exemple pourraient être utilisées pour stimuler de manière adéquate des cellules ganglionnaires exprimant la ChR2 [75–77].
Il est difficile à l’heure actuelle d’anticiper les progrès à venir dans cette voie de recherche tant ils dépendent de nombreux facteurs. Il est clair en revanche que les tests précliniques devront se faire sur des modèles animaux plus proches de l’homme sur le plan visuel que les rongeurs. Des tests sont actuellement en cours chez le chien et le primate non humain afin d’évaluer le bénéfice thérapeutique d’une stratégie basée sur un outil photochimique codé génétiquement. Une discussion sur les critères d’inclusion dans des protocoles de recherche clinique utilisant des techniques optogénétiques a récemment émergé [78], et un premier essai thérapeutique chez l’homme mené par la société RetroSense Therapeutics [79] pourrait démarrer très prochainement aux États-Unis.
Perspectives
La neuromodulation optogénétique offre des perspectives thérapeutiques à moyen terme pour un certain nombre de pathologies du système nerveux clairement identifiées. Dans chacune de ces pathologies, elle est susceptible de se substituer avantageusement aux stimulations électriques en intervenant sur les circuits neuronaux dysfonctionnels avec un degré de spécificité inégalé jusqu’à maintenant. Il est possible que le champ des applications thérapeutiques basées sur l’optogénétique s’élargisse dans le futur, dans le sillage des stratégies de stimulation cérébrale profonde qui sont aujourd’hui testées dans un nombre croissant de maladies neuropsychiatriques (troubles obsessionnels compulsifs sévères [86], dépressions pharmaco-résistantes ou encore syndrome de Gilles de la Tourette), pour la prise en charge de certains types de céphalées (algie vasculaire de la face) ou encore pour lutter contre l’hyperphagie dans le syndrome de Prader-Willi. Dans tous les cas, la neuromodulation optogénétique constitue déjà un atout majeur pour la recherche préclinique, dans le cadre de travaux visant à affiner notre compréhension des mécanismes physio-pathologiques d’un certain nombre de maladies du système nerveux [4, 80].
Malgré des difficultés méthodologiques et des questions d’ordre éthique (fondamentalement similaires à celles déjà soulevées par la thérapie génique), la transposition des techniques de neuromodulation optogénétique à la clinique est déjà sérieusement envisagée. Au cours des sept dernières années, plusieurs start-ups ont été créées dans le but de développer et commercialiser des protocoles de transduction virale et des dispositifs de photostimulation utilisables chez l’homme. Citons par exemple LucCell, qui envisage une stratégie optogénétique pour traiter l’incontinence urinaire, Eos Neuroscience [81], RetroSense Therapeutics [79] et GenSight Biologics [82], dont le but est de traiter des pathologies rétiniennes et enfin Circuit Therapeutics [83], qui compte utiliser la neuromodulation optogénétique pour l’identification de nouvelles cibles thérapeutiques dans une série de maladies neuropsychiatriques. Sur le plan institutionnel, un groupe de chercheurs dans le domaine a été auditionné en juin 2010 par le groupe d’experts sur les dispositifs médicaux de la Commission européenne, signe qu’une réflexion est amorcée sur l’encadrement législatif d’une transposition clinique de cette technique.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Dugue GP, Akemann W, Knopfel T. A comprehensive concept of optogenetics. Prog Brain Res 2012 ; 196 : 1–28. [CrossRef] [PubMed] [Google Scholar]
- Dugue GP, Tricoire L. Principes et applications de l’optogénétique en neuroscience. Med Sci (Paris) 2015 ; 31 : 291–303. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Fenno L, Yizhar O, Deisseroth K. The development and application of optogenetics. Annu Rev Neurosci 2011 ; 34 : 389–412. [CrossRef] [PubMed] [Google Scholar]
- Han MH, Friedman AK. Virogenetic and optogenetic mechanisms to define potential therapeutic targets in psychiatric disorders. Neuropharmacology 2012 ; 62 : 89–100. [CrossRef] [PubMed] [Google Scholar]
- Lozano AM, Lipsman N. Probing and regulating dysfunctional circuits using deep brain stimulation. Neuron 2013 ; 77 : 406–424. [CrossRef] [PubMed] [Google Scholar]
- Lipsman N, Giacobbe P, Lozano AM. Deep brain stimulation in obsessive-compulsive disorder: neurocircuitry and clinical experience. Handb Clin Neurol 2013 ; 116 : 245–250. [CrossRef] [PubMed] [Google Scholar]
- Sankar T, Lipsman N, Lozano AM. Deep brain stimulation for disorders of memory and cognition. Neurotherapeutics 2014 ; 11 : 527–534. [CrossRef] [Google Scholar]
- Lipsman N, Woodside DB, Lozano AM. Neurocircuitry of limbic dysfunction in anorexia nervosa. Cortex 2014 ; 62 : 109–118. [CrossRef] [PubMed] [Google Scholar]
- Mallet L, Schüpbach M, N’Diaye K, et al. Stimulation of subterritories of the subthalamic nucleus reveals its role in the integration of the emotional and motor aspects of behavior. Proc Natl Acad Sci USA 2007 ; 104 : 10661–10666. [CrossRef] [Google Scholar]
- Allen BD, Singer AC, Boyden ES. Principles of designing interpretable optogenetic behavior experiments. Learn Mem 2015 ; 22 : 232–238. [CrossRef] [PubMed] [Google Scholar]
- Ahi YS, Bangari DS, Mittal SK. Adenoviral vector immunity: its implications and circumvention strategies. Curr Gene Ther 2011 ; 11 : 307–320. [CrossRef] [PubMed] [Google Scholar]
- Duale H, Kasparov S, Paton JF, Teschemacher AG. Differences in transductional tropism of adenoviral and lentiviral vectors in the rat brainstem. Exp Physiol 2005 ; 90 : 71–78. [CrossRef] [PubMed] [Google Scholar]
- Christine CW, Starr PA, Larson PS, et al. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 2009 ; 73 : 1662–1669. [CrossRef] [PubMed] [Google Scholar]
- Marks WJ, Jr, Bartus RT, Siffert J, Davis CS, et al. Gene delivery of AAV2-neurturin for Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol 2010 ; 9 : 1164–1172. [CrossRef] [PubMed] [Google Scholar]
- LeWitt PA, Rezai AR, Leehey MA, et al. AAV2-GAD gene therapy for advanced Parkinson’s disease: a double-blind, sham-surgery controlled, randomised trial. Lancet Neurol 2011 ; 10 : 309–319. [CrossRef] [PubMed] [Google Scholar]
- Mukherjee S, Thrasher AJ. Gene therapy for PIDs: progress, pitfalls and prospects. Gene 2013 ; 525 : 174–181. [CrossRef] [PubMed] [Google Scholar]
- Kordower JH, Emborg ME, Bloch J, et al. Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson’s disease. Science 2000 ; 290 : 767–773. [CrossRef] [PubMed] [Google Scholar]
- Palfi S, Gurruchaga JM, Ralph GS, et al. Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: a dose escalation, open-label, phase 1/2 trial. Lancet 2014 ; 383 : 1138–1146. [CrossRef] [PubMed] [Google Scholar]
- Miyashita T, Shao YR, Chung J, et al. Long-term channelrhodopsin-2 (ChR2) expression can induce abnormal axonal morphology and targeting in cerebral cortex. Front Neural Circuits 2013 ; 7 : 8. [PubMed] [Google Scholar]
- Senova YS, et al. Optical stimulation of the cortico-subthalamic pathway using lentiviral vector with retrograde transport properties for CHR-2-eYFP in non-human primates. New Orleans, LA : Society for Neuroscience, 2012. [Google Scholar]
- Gerits A, Farivar R, Rosen BR, et al. Optogenetically induced behavioral and functional network changes in primates. Curr Biol 2012 ; 22 : 1722–1726. [CrossRef] [PubMed] [Google Scholar]
- Klapoetke NC, Murata Y, Kim SS, et al. Independent optical excitation of distinct neural populations. Nat Methods 2014 ; 11 : 338–346. [CrossRef] [PubMed] [Google Scholar]
- Lin JY, Knutsen PM, Muller A, et al. ReaChR: a red-shifted variant of channelrhodopsin enables deep transcranial optogenetic excitation. Nat Neurosci 2013 ; 16 : 1499–1508. [Google Scholar]
- Chuong AS, Miri ML, Busskamp V, et al. Noninvasive optical inhibition with a red-shifted microbial rhodopsin. Nat Neurosci 2014 ; 17 : 1123–1129. [Google Scholar]
- Berndt A, Yizhar O, Gunaydin LA, et al. Bi-stable neural state switches. Nat Neurosci 2009 ; 12 : 229–234. [CrossRef] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 2011 ; 477 : 171–178. [CrossRef] [PubMed] [Google Scholar]
- Han X., Qian X, Bernstein JG, et al. Millisecond-timescale optical control of neural dynamics in the nonhuman primate brain. Neuron 2009 ; 62 : 191–198. [CrossRef] [PubMed] [Google Scholar]
- Han X, Chow BY, Zhou H, et al. A high-light sensitivity optical neural silencer: development and application to optogenetic control of non-human primate cortex. Front Syst Neurosci 2011 ; 5 : 18. [PubMed] [Google Scholar]
- Diester I, Kaufman MT, Mogri M, et al. An optogenetic toolbox designed for primates. Nat Neurosci 2011 ; 14 : 387–397. [CrossRef] [PubMed] [Google Scholar]
- Cavanaugh J, Monosov IE, McAlonan K, et al. Optogenetic inactivation modifies monkey visuomotor behavior. Neuron 2012 ; 76 : 901–907. [CrossRef] [PubMed] [Google Scholar]
- Lang AE, Lozano AM. Parkinson’s disease. First of two parts. N Engl J Med 1998 ; 339 : 1044–1053. [CrossRef] [PubMed] [Google Scholar]
- Lang AE, Lozano AM. Parkinson’s disease. Second of two parts. N Engl J Med 1998 ; 339 : 1130–1143. [CrossRef] [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PR, et al. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 2010 ; 466 : 622–626. [CrossRef] [PubMed] [Google Scholar]
- Prashanth LK, Fox S, Meissner WG. L-Dopa-induced dyskinesia-clinical presentation, genetics, and treatment. Int Rev Neurobiol 2011 ; 98 : 31–54. [CrossRef] [PubMed] [Google Scholar]
- Benabid AL, Pollak P, Gross C, et al. Acute and long-term effects of subthalamic nucleus stimulation in Parkinson’s disease. Stereotact Funct Neurosurg 1994 ; 62 : 76–84. [Google Scholar]
- Carron R, Chabardes S, Hammond C. Les mécanismes d’action de la simulation cérébrale à haute fréquence. Neurochirurgie 2012 ; 58 : 209–217. [CrossRef] [PubMed] [Google Scholar]
- Gradinaru V, Mogri M, Thompson KR, et al. Optical deconstruction of parkinsonian neural circuitry. Science 2009 ; 324 : 354–359. [CrossRef] [PubMed] [Google Scholar]
- Drouot X, Oshino S, Jarraya B, et al. Functional recovery in a primate model of Parkinson’s disease following motor cortex stimulation. Neuron 2004 ; 44 : 769–778. [CrossRef] [PubMed] [Google Scholar]
- Lefaucheur JP, Drouot X, Von Raison F, et al. Improvement of motor performance and modulation of cortical excitability by repetitive transcranial magnetic stimulation of the motor cortex in Parkinson’s disease. Clin Neurophysiol 2004 ; 115 : 2530–2541. [CrossRef] [PubMed] [Google Scholar]
- Mendes Martins V, Coste J, Derost P, et al. Complications chirurgicales de la stimulation cérébrale profonde : expérience clinique à propos de 184 cas. Neurochirurgie 2012; 58 : 219–224. [CrossRef] [PubMed] [Google Scholar]
- Picot MC, Baldy-Moulinier M, Daurès JP, et al. The prevalence of epilepsy and pharmacoresistant epilepsy in adults: a population-based study in a Western European country. Epilepsia 2008 ; 49 : 1230–1238. [CrossRef] [PubMed] [Google Scholar]
- Laxpati NG, Kasoff WS, Gross RE. Deep brain stimulation for the treatment of epilepsy: circuits, targets, and trials. Neurotherapeutics 2014 ; 11 : 508–526. [CrossRef] [Google Scholar]
- Fisher R, Salanova V, Witt T, et al. Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia 2010 ; 51 : 899–908. [CrossRef] [PubMed] [Google Scholar]
- Tonnesen J, Sørensen AT, Deisseroth K, et al. Optogenetic control of epileptiform activity. Proc Natl Acad Sci USA 2009 ; 106 : 12162–12167. [CrossRef] [Google Scholar]
- Wykes RC, Heeroma JH, Mantoan L, et al. Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci Transl Med 2012; 4 : 161ra152. [CrossRef] [PubMed] [Google Scholar]
- Berglind F, Ledri M, Sørensen AT, et al. Optogenetic inhibition of chemically induced hypersynchronized bursting in mice. Neurobiol Dis 2014 ; 65 : 133–141. [CrossRef] [PubMed] [Google Scholar]
- Sukhotinsky I, Chan AM, Ahmed OJ, et al. Optogenetic delay of status epilepticus onset in an in vivo rodent epilepsy model. PLoS One 2013 ; 8 : e62013. [CrossRef] [PubMed] [Google Scholar]
- Ledri M, Madsen MG, Nikitidou L, et al. Global optogenetic activation of inhibitory interneurons during epileptiform activity. J Neurosci 2014 ; 34 : 3364–3377. [CrossRef] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, et al. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science 2002 ; 298 : 1418–1421. [CrossRef] [PubMed] [Google Scholar]
- Wozny C, Kivi A, Lehmann TN, et al. Comment on: on the origin of interictal activity in human temporal lobe epilepsy in vitro. Science 2003 ; 301 : 463. [CrossRef] [Google Scholar]
- Paz JZ, Davidson TJ, Frechette ES, et al. Closed-loop optogenetic control of thalamus as a tool for interrupting seizures after cortical injury. Nat Neurosci 2013 ; 16 : 64–70. [CrossRef] [PubMed] [Google Scholar]
- Krook-Magnuson E, Armstrong C, Oijala M, Soltesz I. On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat Commun 2013 ; 4 : 1376. [CrossRef] [PubMed] [Google Scholar]
- Mendes HF, van der Spuy J, Chapple JP, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med 2005 ; 11 : 177–185. [CrossRef] [PubMed] [Google Scholar]
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet 2006 ; 368 : 1795–1809. [CrossRef] [PubMed] [Google Scholar]
- Fahim AT, Daiger SP, Weleber RG Retinitis pigmentosa overview. Seattle (WA) : GeneReviews, 2000. [Google Scholar]
- Maghami MH, Sodagar AM, Lashay A, et al. Visual prostheses: the enabling technology to give sight to the blind. J Ophthalmic Vis Res 2014 ; 9 : 494–505. [CrossRef] [PubMed] [Google Scholar]
- Humayun MS, Dorn JD, da Cruz L, et al. Interim results from the international trial of second sight’s visual prosthesis. Ophthalmology 2012 ; 119 : 779–788. [CrossRef] [PubMed] [Google Scholar]
- Uy HS, Chan PS, Cruz FM. Stem cell therapy: a novel approach for vision restoration in retinitis pigmentosa. Med Hypothesis Discov Innov Ophthalmol 2013 ; 2 : 52–55. [PubMed] [Google Scholar]
- Tao W. Application of encapsulated cell technology for retinal degenerative diseases. Expert Opin Biol Ther 2006 ; 6 : 717–726. [CrossRef] [PubMed] [Google Scholar]
- Petrs-Silva H, Linden R. Advances in gene therapy technologies to treat retinitis pigmentosa. Clin Ophthalmol 2014 ; 8 : 127–136. [PubMed] [Google Scholar]
- Sahel JA, Roska B. Gene therapy for blindness. Annu Rev Neurosci 2013 ; 36 : 467–488. [CrossRef] [PubMed] [Google Scholar]
- Boye SE, Boye SL, Lewin AS, Hauswirth WW. A comprehensive review of retinal gene therapy. Mol Ther 2013 ; 21 : 509–519. [CrossRef] [PubMed] [Google Scholar]
- Dalkara D, Sahel JA. Gene therapy for inherited retinal degenerations. CR Biol 2014 ; 337 : 185–192. [CrossRef] [Google Scholar]
- Stieger K, Cronin T, Bennett J, Rolling F. Adeno-associated virus mediated gene therapy for retinal degenerative diseases. Methods Mol Biol 2011 ; 807 : 179–218. [CrossRef] [PubMed] [Google Scholar]
- Bi A, Cui J, Ma YP, et al. Ectopic expression of a microbial-type rhodopsin restores visual responses in mice with photoreceptor degeneration. Neuron 2006 ; 50 : 23–33. [CrossRef] [PubMed] [Google Scholar]
- Tomita H, Sugano E, Fukazawa Y, et al. Visual properties of transgenic rats harboring the channelrhodopsin-2 gene regulated by the thy-1.2 promoter. PLoS One 2009 ; 4 : e7679. [CrossRef] [PubMed] [Google Scholar]
- Thyagarajan S, van Wyk M, Lehmann K, et al. Visual function in mice with photoreceptor degeneration and transgenic expression of channelrhodopsin 2 in ganglion cells. J Neurosci 2010 ; 30 : 8745–8758. [CrossRef] [PubMed] [Google Scholar]
- Lin B, Koizumi A, Tanaka N, et al. Restoration of visual function in retinal degeneration mice by ectopic expression of melanopsin. Proc Natl Acad Sci USA 2008 ; 105 : 16009–16014. [CrossRef] [Google Scholar]
- Caporale N, Kolstad KD, Lee T, et al. LiGluR restores visual responses in rodent models of inherited blindness. Mol Ther 2011 ; 19 : 1212–1219. [Google Scholar]
- Lagali PS, Balya D, Awatramani GB, et al. Light-activated channels targeted to ON bipolar cells restore visual function in retinal degeneration. Nat Neurosci 2008 ; 11 : 667–675. [CrossRef] [PubMed] [Google Scholar]
- Cronin T., Vandenberghe LH, Hantz P, et al. Efficient transduction and optogenetic stimulation of retinal bipolar cells by a synthetic adeno-associated virus capsid and promoter. EMBO Mol Med 2014 ; 6 : 1175–1190. [CrossRef] [PubMed] [Google Scholar]
- Mace E, Vandenberghe LH, Hantz P, et al. Targeting channelrhodopsin-2 to ON-bipolar cells with vitreally administered AAV restores ON and OFF visual responses in blind mice. Mol Ther 2014 ; 23 : 7–16. [Google Scholar]
- Doroudchi MM, Greenberg KP, Liu J, et al. Virally delivered channelrhodopsin-2 safely and effectively restores visual function in multiple mouse models of blindness. Mol Ther 2011 ; 19 : 1220–1229. [CrossRef] [PubMed] [Google Scholar]
- Busskamp V, Duebel J, Balya D, et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science 2010 ; 329 : 413–417. [CrossRef] [PubMed] [Google Scholar]
- Reutsky-Gefen I, Golan L, Farah N, et al. Holographic optogenetic stimulation of patterned neuronal activity for vision restoration. Nat Commun 2013 ; 4 : 1509. [CrossRef] [PubMed] [Google Scholar]
- Nirenberg S, Pandarinath C. Retinal prosthetic strategy with the capacity to restore normal vision. Proc Natl Acad Sci USA 2012 ; 109 : 15012–15017. [CrossRef] [Google Scholar]
- Barrett JM, Berlinguer-Palmini R, Degenaar P. Optogenetic approaches to retinal prosthesis. Vis Neurosci 2014 ; 31 : 345–354. [CrossRef] [PubMed] [Google Scholar]
- Jacobson SG, Sumaroka A, Luo X, Cideciyan AV. Retinal optogenetic therapies: clinical criteria for candidacy. Clin Genet 2013 ; 84 : 175–182. [CrossRef] [PubMed] [Google Scholar]
- RetroSense Therapeutics. Available from: http://www.retro-sense.com/. [Google Scholar]
- McDevitt RA, Reed SJ, Britt JP. Optogenetics in preclinical neuroscience and psychiatry research: recent insights and potential applications. Neuropsychiatr Dis Treat 2014 ; 10 : 1369–1379. [PubMed] [Google Scholar]
- Eos Neuroscience. Available from: http://www.eosneuroscience.com/. [Google Scholar]
- GenSight Biologics. Available from: http://www.gensight-biologics.com. [Google Scholar]
- Circuit Therapeutics. Available from: http://circuittx.com/. [Google Scholar]
- Redgrave P, Rodriguez M, Smith Y, et al. Goal-directed and habitual control in the basal ganglia: implications for Parkinson’s disease. Nat Rev Neurosci 2010 ; 11 : 760–772. [CrossRef] [PubMed] [Google Scholar]
- Dalkara D. La conception de vecteurs adaptés à la thérapie génique oculaire. Med Sci (Paris) 2015; 31 (sous presse). [Google Scholar]
- Flores Alves Dos Santos J, Mallet L. Le trouble obsessionnel compulsif. Med Sci (Paris) 2013 ; 29 : 1111–1116. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Photosensibilisation de cellules nerveuses par transduction virale. A. Un transgène (ici codant la channelrhodopsine-2) est encapsidé dans des particules virales qui sont ensuite introduites dans une région cérébrale donnée par injection stéréotaxique. B. Pour des expériences de neuromodulation optogénétique sur des animaux en situation comportementale, un segment de fibre optique fixé dans un connecteur miniature peut être implanté sur la tête de l’animal. Lors de l’expérience, ce connecteur est relié à une source de lumière par l’intermédiaire d’un câble optique. C. La lumière délivrée par la fibre optique n’agit que sur les neurones photosensibilisés (ici en vert), c’est-à-dire ceux exprimant la protéine codée par le transgène. |
| Dans le texte | |
|
Figure 2. Exemples de dispositifs cliniques utilisés actuellement pour les injections et stimulations intracrâniennes. A. Dispositif d’injection stéréotaxique de vecteurs viraux. B. Électrodes de stimulation implantées dans le noyau sous-thalamique d’un patient parkinsonien et révélées par radiographie. |
| Dans le texte | |
|
Figure 3. Implication du faisceau cortico-sous-thalamique dans le mécanisme d’action de la stimulation du noyau sous-thalamique chez le patient parkinsonien. A. Photographie (à gauche) et schéma (à droite) représentant une coupe coronale de cerveau humain. Une partie des ganglions de la base sont représentés en couleur sur le schéma. Le code couleur des différents noyaux est identique à celui du diagramme en B. Le faisceau cortico-sous-thalamique (voie hyperdirecte) est représenté en vert. B. Diagramme de connectivité simplifié des ganglions de la base représentant la voie directe, la voie indirecte, ainsi que la voie hyperdirecte. Chez la souris, la photostimulation (flèche bleue) sélective des afférences de la voie hyperdirecte dans le noyau sous-thalamique suffit à reproduire les effets antiparkinsoniens de la stimulation électrique du noyau sous-thalamique [37]. D1 et D2 : neurones striataux exprimant les récepteurs dopaminergiques D1 et D2 respectivement ; GPe : globus pallidus externe ; GPi : globus pallidus interne ; NST : noyau sous-thalamique ; SNc : substance noire pars compacta ; SNr : substance noire pars reticulata (adapté de [84]). |
| Dans le texte | |
|
Figure 4. Stratégies optogénétiques pour le traitement de l’épilepsie. A-B. Deux options sont possibles : inhiber les neurones excitateurs impliqués dans la genèse ou la propagation de l’activité ictale (A) , ou exciter les interneurones inhibiteurs locaux (B) , par expression spécifique de pompes hyperpolarisantes (telles que la NpHR) ou d’outils dépolarisants (tels que la ChR2) respectivement. C. Principe de la neuromodulation optogénétique en boucle fermée, qui permet de n’illuminer le tissu nerveux que lorsqu’une crise est détectée. EEG : électro-encéphalogramme. D. Exemples de tracés électrophysiologiques démontrant l’efficacité de l’inhibition optogénétique en boucle fermée dans un modèle d’épilepsie du lobe temporal chez la souris. La ligne verte pointillée indique le moment où l’algorithme détecte l’arrivée d’une crise. L’activité épileptiforme peut être stoppée lorsque cet algorithme déclenche l’illumination du tissu. Reproduit d’après [52]. |
| Dans le texte | |
|
Figure 5. Différentes options pour le traitement optogénétique des rétinopathies pigmentaires. A. Schéma anatomique de l’œil humain. Les vecteurs viraux peuvent être injectés dans l’humeur vitrée (injection intra-vitréenne) ou sous la rétine. Les injections sous-rétiniennes présentent l’inconvénient d’endommager mécaniquement un tissu déjà fragilisé par le processus de dégénérescence, et de ne pouvoir atteindre que des surfaces restreintes de rétine. B. Schéma simplifié du circuit rétinien, montrant les deux principaux types de circuits (ON et OFF) en aval des photorécepteurs à cônes. Plusieurs types neuronaux ne sont pas représentés (cellules amacrines et cellules horizontales). C. Stade avancé de dégénérescence rétinienne dans lequel seuls certains photorécepteurs à cônes subsistent sous la forme de cellules atrophiées (cônes dormants). La rephotosensibilisation du circuit peut s’appuyer sur l’expression de la ChR2 dans les cellules ganglionnaires (accessibles par voie intra-vitréenne à l’aide d’AAV uniquement), par expression de la ChR2 dans les cellules bipolaires de type ON ou de NpHR dans les cônes dormants (types cellulaires accessibles par injection sous-rétinienne de plusieurs classes de virus, et par injection intra-vitréenne d’AAV présentant un fort pouvoir de diffusion). |
| Dans le texte | |
Les statistiques affichées correspondent au cumul d'une part des vues des résumés de l'article et d'autre part des vues et téléchargements de l'article plein-texte (PDF, Full-HTML, ePub... selon les formats disponibles) sur la platefome Vision4Press.
Les statistiques sont disponibles avec un délai de 48 à 96 heures et sont mises à jour quotidiennement en semaine.
Le chargement des statistiques peut être long.