")
")
| Issue |
Med Sci (Paris)
Volume 42, Number 3, Mars 2026
|
|
|---|---|---|
| Page(s) | 230 - 233 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2026025 | |
| Published online | 20 March 2026 | |
La production intestinale d’hepcidine réduit l’absorption du fer
Une nouvelle arme pour lutter contre les maladies de surcharge en fer ?
Intestinal production of hepcidin reduces iron absorption: a new weapon against iron overload disorders?
1
Université Paris Cité, CNRS, Inserm U1016, Institut Cochin, Paris, France
2
Initiatives IdEx Globule rouge d’excellence (InIdex globules rouges-Ex), Université Paris Cité, Paris, France
3
UMR Physiologie, environnement et génétique pour l’animal et les systèmes d’élevage (PEGASE), INRAE, Institut Agro, Saint Gilles, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
**
This email address is being protected from spambots. You need JavaScript enabled to view it.
***
This email address is being protected from spambots. You need JavaScript enabled to view it.
Le fer est un micronutriment nécessaire à la vie. Il joue un rôle clé dans de nombreux processus cellulaires (métabolisme bioénergétique, réplication de l’ADN, régulation de l’expression des gènes, etc.) et permet l’oxygénation des tissus par sa contribution au noyau hème de l’hémoglobine des globules rouges du sang. Les propriétés biologiques du fer reposent en partie sur sa capacité à catalyser les réactions d’oxydo-réduction, ce qui peut également le rendre toxique pour la cellule lorsqu’il est présent en excès. En effet, il favorise alors, par la réaction de Fenton1, la production d’espèces réactives de l’oxygène délétères.
L’homéostasie du fer chez les mammifères est contrôlée par l’axe hepcidineferroportine. L’hepcidine (codée par le gène HAMP) est l’hormone du fer. Elle est hyposidérémiante. Son rôle principal est d’empêcher l’excès de fer dans l’organisme, et d’éviter ainsi qu’une production excessive d’espèces réactives de l’oxygène n’endommage les tissus. L’activité de l’hepcidine repose sur sa capacité à provoquer la dégradation de la ferroportine, le seul exporteur de fer connu à ce jour, en se liant à cette protéine de transport et en induisant sa dégradation dans les lysosomes. Ainsi, l’hepcidine module l’absorption alimentaire intestinale de fer par les entérocytes et son recyclage par les macrophages (principalement ceux de la rate, qui recyclent massivement le fer provenant de la phagocytose des globules rouges sénescents) (Figure 1). Des souris mutantes privées d’hepcidine par invalidation du gène Hamp, ayant perdu cette capacité à freiner l’absorption et le recyclage du fer, voient leurs tissus se charger progressivement en fer [1]. Ce phénotype mime celui d’une maladie génétique humaine : l’hémochromatose héréditaire, caractérisée par une surcharge en fer systémique et tissulaire. Cette maladie est due à un déficit de production d’hepcidine, et sa gravité dépend de la quantité résiduelle de l’hormone. La forme la plus fréquente est causée par la mutation du gène HFE, qui code un facteur impliqué dans la régulation de la production d’hepcidine par le foie [2]. Au contraire, des souris transgéniques dont le foie produit des quantités anormalement élevées d’hepcidine deviennent anémiques par manque de fer [3]. Chez l’Homme, un tel phénotype de surexpression de l’hepcidine est retrouvé dans une forme rare d’anémie génétique (iron refractory iron deficient anemia, IRIDA), due à la mutation du gène TMPRSS6 (Transmembrane serine protease 6) codant la matriptase 2, un régulateur négatif de l’hepcidine [4].
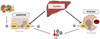 |
Figure 1 Métabolisme du fer et son contrôle par l’hepcidine. Le fer alimentaire est absorbé par les entérocytes du duodénum afin de compenser les pertes non spécifiques de fer dues aux saignements ou à la desquamation des cellules intestinales. Le fer est importé au pôle apical de l’entérocyte par le transporteur DMT1 (divalent metal transporter 1), et il est exporté à son pôle basolatéral par la ferroportine (FPN). Des systèmes de régulation contrôlent l’absorption intestinale du fer afin d’éviter son accumulation dans les tissus. La quantité de fer alimentaire absorbée est faible (de l’ordre de 1 à 2 mg par jour), et l’essentiel du fer présent dans le plasma provient des macrophages (en particulier ceux de la rate) qui phagocytent les érythrocytes sénescents et remettent à disposition dans la circulation le fer de l’hémoglobine qu’ils contiennent (en moyenne, 20 à 25 mg de fer par jour). Dans le plasma, le fer est lié à la transferrine, et il est principalement utilisé pour la production d’hémoglobine dans les cellules érythroïdes de la moelle osseuse. L’hepcidine présente dans la circulation sanguine provient très majoritairement du foie. Elle agit sur la ferroportine, dont elle induit la dégradation. Elle a donc un effet hyposidérémiant, et permet d’éviter tout excès toxique de la quantité de fer présente dans l’organisme. |
L’hepcidine est produite très majoritairement par le foie. Ainsi, des souris chez lesquelles seule la synthèse hépatique d’hepcidine a été supprimée présentent un phénotype de surcharge en fer identique à celui des souris totalement dépourvues d’hepcidine, ce qui indique qu’aucun autre tissu n’est capable de compenser le déficit d’hepcidine d’origine hépatique [5]. Toutefois, les résultats de plusieurs études indiquent que l’hepcidine peut également être produite par d’autres cellules que les hépatocytes, dans des situations pathologiques [6]. Nous avons notamment montré que dans le psoriasis, une maladie inflammatoire chronique, la surexpression d’hepcidine dans les kératinocytes était responsable de l’accumulation de fer dans la peau (par un effet local de l’hormone sur la dégradation de la ferroportine des kératinocytes), et des signes cutanés de la maladie (prolifération des kératinocytes, épaississement de la peau, et inflammation locale avec recrutement des granulocytes neutrophiles) [7].
Plus récemment, c’est un autre tissu qui a retenu notre attention à la suite de différentes études montrant la synthèse ectopique de l’hormone dans l’intestin en situation pathologique [8, 9]. Pour comprendre le rôle de cette production intestinale d’hepcidine dans le métabolisme du fer, nous avons produit un modèle murin transgénique surexprimant l’hepcidine spécifiquement dans les cellules épithéliales de l’intestin (souris Hamp1KI-Int) [10]. Dès l’âge de trois semaines, ces souris présentent une anémie par carence en fer, avec dans le plasma sanguin, une diminution de la concentration en fer, du coefficient de saturation de la transferrine, et de la ferritine (un marqueur sérique témoin de la charge en fer). Elles ont par ailleurs une perte de poils, autre signe caractéristique du déficit en fer. La preuve du rôle de la surexpression intestinale d’hepcidine dans le phénotype de ces souris a été apportée par la mesure de l’absorption intestinale du fer isotopique 57Fe, qui était nettement diminuée en comparaison de celle des souris témoins [10].
Mais alors, où et comment agit l’hepcidine produite en excès dans l’intestin pour réduire l’absorption du fer ? Cette hepcidine agit-elle en tant qu’hormone, après son passage dans la circulation sanguine, ou bien par une éventuelle sécrétion dans la lumière intestinale ? Étonnamment, nous n’avons pas détecté d’effet hormonal de l’hepcidine transgénique sur la dégradation de la ferroportine, située au pôle basolatéral des entérocytes, alors même que cette hepcidine est présente dans le sang des souris Hamp1KI-Int. Cependant, nous avons montré, chez ces souris, la présence d’hepcidine dans la lumière intestinale, ce qui suggérait un possible effet sur la protéine DMT1 (divalent metal transporter 1), l’importeur du fer situé au pôle apical des entérocytes (voir Figure 1). Effectivement, l’analyse a montré une franche réduction de la quantité de DMT1 dans les entérocytes des souris Hamp1KI-Int, qui explique vraisemblablement l’inhibition de l’absorption du fer chez ces souris et leur anémie [10]. Cette action de l’hepcidine sur DMT1, qui avait déjà été suggérée en se fondant sur des résultats obtenus in vitro et ex vivo [11, 12], vient ainsi d’être confirmée in vivo (Figure 2).
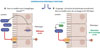 |
Figure 2 Conséquences d’une surproduction intestinale d’hepcidine. C’est principalement l’hepcidine d’origine hépatique qui contribue à l’homéostasie du fer, mais dans des conditions pathologiques, l’hepcidine peut également être produite par l’intestin. A. En créant des souris transgéniques surexprimant l’hepcidine dans les entérocytes (souris Hamp1KI-int), nous avons montré qu’elle est alors sécrétée dans la lumière intestinale au pôle apical des entérocytes, et qu’elle inhibe directement l’absorption intestinale du fer en agissant sur l’importeur du fer DMT1, ce qui conduit à un phénotype de déficience en fer et d’anémie. B. Nous avons également utilisé une bactérie probiotique (L. lactis) recombinante pour délivrer directement l’hepcidine dans la lumière intestinale. Cette stratégie a permis de réduire la surcharge en fer dans un modèle murin d’hémochromatose (souris KO Hepc). |
Afin d’évaluer la capacité de l’hepcidine présente dans la lumière intestinale à prévenir la surcharge en fer chez les souris invalidées pour le gène Hamp, modèle murin d’hémochromatose, nous avons choisi, pour produire cette hepcidine, d’utiliser une bactérie probiotique. Une souche recombinante de Lactobacillus incluant le gène de l’hepcidine (LAB-Hepc) a été créée, et son efficacité pour la production d’hepcidine a été validée in vitro. Des souris invalidées pour Hamp ont alors reçu le probiotique LAB-Hepc par gavage pendant un mois. Alors que les souris non traitées par le probiotique ont développé, comme attendu, une surcharge en fer, nous avons constaté une réduction spectaculaire de la quantité de fer dans l’organisme des souris traitées, en particulier dans le foie, ce qui atteste le potentiel thérapeutique du probiotique LAB-Hepc [10] (Figure 2).
Grâce au modèle des souris transgéniques surexprimant l’hepcidine dans les entérocytes, nous avons donc apporté la preuve in vivo d’un puissant rôle freinateur de l’hepcidine sécrétée dans la lumière intestinale sur l’absorption du fer alimentaire, en agissant sur son transporteur DMT1 au pôle apical des entérocytes, par un mécanisme que nous cherchons aujourd’hui à préciser (Figure 2). De plus, le succès de l’utilisation du probiotique LAB-Hepc chez la souris pour réduire la charge en fer ouvre une perspective de thérapie innovante et non invasive, non seulement pour l’hémochromatose, mais vraisemblablement aussi pour les autres maladies de surcharge en fer, notamment celles comportant une dysérythropoïèse par altération de la structure ou dysfonctionnement de l’hémoglobine (drépanocytose, thalassémies, ou anémies sidéroblastiques), ou par altération de la production des globules rouges, comme dans la myélodysplasie.
Les résultats d’un nombre croissant d’études montrent que l’hepcidine peut être synthétisée dans d’autres organes que le foie, l’intestin, ou la peau (rein, tissu adipeux, cœur, estomac, pancréas, cerveau, etc.), ainsi que dans les cellules immunitaires (macrophages, cellules dendritiques, granulocytes neutrophiles, lymphocytes T) [6]. Elle pourrait, par une action autocrine ou paracrine, y activer les processus cellulaires dépendant du fer en augmentant la dégradation de la ferroportine. Se pose alors la question du mécanisme par lequel le fer disponible dans un organe ou un tissu est réparti entre ses différents types cellulaires, alors que leurs besoins peuvent différer selon le contexte physiologique ou physiopathologique (infection, inflammation, cancer, etc.), et que les nombreuses protéines dont l’activité nécessite la présence du fer ont vraisemblablement des sensibilités variables à la privation ou à la surcharge en fer.
Remerciements
Nous remercions les membres du laboratoire dirigé par Philippe Langella (laboratoire MICALIS-Univ. Paris-Saclay/INRAE/AgroParisTech) pour la production de la souche recombinante de Lactobacillus.
Liens d’intérêt
Les auteures déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article
Références
- Lesbordes-Brion JC, Viatte L, Bennoun M, et al. Targeted disruption of the hepcidin 1 gene results in severe hemochromatosis. Blood 8 ; 108 : 1402–5. [Google Scholar]
- Girelli D, Marchi G, Busti F. Diagnosis and management of hereditary hemochromatosis: lifestyle modification, phlebotomy, and blood donation. Hematology Am Soc Hematol Educ Program 2024 ; 2024 : 434–42. [Google Scholar]
- Nicolas G, Bennoun M, Porteu A, et al. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci USA 2002 ; 99 : 4596–601. [CrossRef] [PubMed] [Google Scholar]
- Finberg KE, Heeney MM, Campagna DR, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat Genet 2008 ; 40 : 569–71. [Google Scholar]
- Zumerle S, Mathieu JR, Delga S, et al. Targeted disruption of hepcidin in the liver recapitulates the hemochromatotic phenotype. Blood 2014 ; 123 : 3646–50. [Google Scholar]
- Lakhal-Littleton S, Peyssonnaux C. Hepcidin and tissue-specific iron regulatory networks. Adv Exp Med Biol 2025 ; 1480: 89–102. [Google Scholar]
- Abboud E, Chrayteh D, Boussetta N, et al. Skin hepcidin initiates psoriasiform skin inflammation via Fe-driven hyperproliferation and neutrophil recruitment. Nat Commun 2024 ; 15 : 6718. [Google Scholar]
- Schwartz AJ, Goyert JW, Solanki S, et al. Hepcidin sequesters iron to sustain nucleotide metabolism and mitochondrial function in colorectal cancer epithelial cells. Nat Metab 2021 ; 3 : 969–82. [Google Scholar]
- Gotardo EM, Ribeiro G, Clemente TR, et al. Hepcidin expression in colon during trinitrobenzene sulfonic acid-induced colitis in rats. World J Gastroenterol 2014 ; 20 : 4345–52. [Google Scholar]
- Falabrègue M, Aurrand C, Cazaulon L, et al. Intestinal hepcidin overexpression promotes iron deficiency anemia and counteracts iron overload via DMT1 downregulation. Blood 2025 ; 146 : 2863–9. [Google Scholar]
- Brasse-Lagnel C, Karim Z, Letteron P, et al. Intestinal DMT1 cotransporter is down-regulated by hepcidin via proteasome internalization and degradation. Gastroenterology 2011 ; 140 : 1261–71.e1. [Google Scholar]
- Chaston T, Chung B, Mascarenhas M, et al. Evidence for differential effects of hepcidin in macrophages and intestinal epithelial cells. Gut 2008 ; 57 : 374–82. [Google Scholar]
La réaction de Fenton est un processus d’oxydation avancée consistant à amorcer des réactions de décomposition du peroxyde d’hydrogène (H202) par des ions métalliques (en particulier Fe2+) afin de produire des espèces chimiques radicalaires, très réactives.
© 2026 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Liste des figures
|
Figure 1 Métabolisme du fer et son contrôle par l’hepcidine. Le fer alimentaire est absorbé par les entérocytes du duodénum afin de compenser les pertes non spécifiques de fer dues aux saignements ou à la desquamation des cellules intestinales. Le fer est importé au pôle apical de l’entérocyte par le transporteur DMT1 (divalent metal transporter 1), et il est exporté à son pôle basolatéral par la ferroportine (FPN). Des systèmes de régulation contrôlent l’absorption intestinale du fer afin d’éviter son accumulation dans les tissus. La quantité de fer alimentaire absorbée est faible (de l’ordre de 1 à 2 mg par jour), et l’essentiel du fer présent dans le plasma provient des macrophages (en particulier ceux de la rate) qui phagocytent les érythrocytes sénescents et remettent à disposition dans la circulation le fer de l’hémoglobine qu’ils contiennent (en moyenne, 20 à 25 mg de fer par jour). Dans le plasma, le fer est lié à la transferrine, et il est principalement utilisé pour la production d’hémoglobine dans les cellules érythroïdes de la moelle osseuse. L’hepcidine présente dans la circulation sanguine provient très majoritairement du foie. Elle agit sur la ferroportine, dont elle induit la dégradation. Elle a donc un effet hyposidérémiant, et permet d’éviter tout excès toxique de la quantité de fer présente dans l’organisme. |
| Dans le texte | |
|
Figure 2 Conséquences d’une surproduction intestinale d’hepcidine. C’est principalement l’hepcidine d’origine hépatique qui contribue à l’homéostasie du fer, mais dans des conditions pathologiques, l’hepcidine peut également être produite par l’intestin. A. En créant des souris transgéniques surexprimant l’hepcidine dans les entérocytes (souris Hamp1KI-int), nous avons montré qu’elle est alors sécrétée dans la lumière intestinale au pôle apical des entérocytes, et qu’elle inhibe directement l’absorption intestinale du fer en agissant sur l’importeur du fer DMT1, ce qui conduit à un phénotype de déficience en fer et d’anémie. B. Nous avons également utilisé une bactérie probiotique (L. lactis) recombinante pour délivrer directement l’hepcidine dans la lumière intestinale. Cette stratégie a permis de réduire la surcharge en fer dans un modèle murin d’hémochromatose (souris KO Hepc). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.