")
")
| Issue |
Med Sci (Paris)
Volume 41, Number 1, Janvier 2025
|
|
|---|---|---|
| Page(s) | 33 - 39 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2024191 | |
| Published online | 31 January 2025 | |
Une approche CRISPR/Cas pour traiter les β-hémoglobinopathies
A CRISPR/Cas approach to β-haemoglobinopathies
Institut Imagine, Inserm UMR1163, université Paris Cité, Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
**
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les β-hémoglobinopathies sont des anémies génétiques graves dues à des mutations affectant l’hémoglobine adulte. Pour y remédier, le système CRISPR/Cas9 a été utilisé pour modifier génétiquement les cellules souches/progénitrices hématopoïétiques des patients ex vivo, et réactiver l’expression de l’hémoglobine fœtale dans la lignée érythroïde. Plus de 70 patients atteints de β-thalassémie ou de drépanocytose ont reçu la thérapie Casgevy®. La plupart de ces patients ont présenté une amélioration notable de leur phénotype clinique, avec une grande efficacité d’édition et des taux d’hémoglobine normaux ou presque. Bien que la sécurité et l’efficacité à long terme doivent encore être évaluées, des stratégies sont en développement pour améliorer les résultats, réduire la génotoxicité potentielle et diminuer les coûts.
Abstract
Beta-haemoglobinopathies are severe genetic anemias caused by mutations that affect adult haemoglobin production. Many therapeutic approaches aim to reactivate the expression of the fetal hemoglobin genes. To this end, the CRISPR/Cas9 system has recently been used to genetically modify patients’ hematopoietic stem/progenitor cells ex vivo and reactivate fetal hemoglobin expression in their erythroid progeny. More than 70 patients with severe β-thalassemia and sickle cell disease have been treated with the Casgevy® therapy. Most have achieved a significant improvement of clinical phenotype, with high editing efficiency in hematopoietic cells associated with normal or near normal hemoglobin levels. While the long-term safety and efficacy of this powerful approach still need to be evaluated, new strategies are being developed to further improve therapeutic outcomes, reduce potential genotoxicity and lower the costs of therapy.
© 2025 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (© Inserm/Claude Féo)
Les β-hémoglobinopathies
L’hémoglobine (Hb) est la protéine majoritaire contenue dans les globules rouges et permet le transport de l’oxygène vers les différents tissus et organes du corps. Elle est constituée de deux chaînes de globine de type α (globine-α) et de deux chaînes de globine de type β (globine-β adulte ou globine-γ fœtale) (Figure 1). Les β-hémoglobinopathies, telles que la drépanocytose et la β-thalassémie, sont des maladies génétiques autosomiques récessives causées par des mutations affectant la production de la chaîne de globine-β du tétramère d’hémoglobine adulte (HbA). Dans la drépanocytose, une mutation ponctuelle (rs334 A>T) entraîne la substitution de l’acide glutamique par la valine dans la chaîne de globine-β (globine-βS), ce qui provoque la polymérisation de l’hémoglobine falciforme (HbS, sickle hemoglobin en anglais) et la formation de globules rouges en forme de faucille [1, 2] (Figure 1). Ces cellules altérées obstruent les petits vaisseaux, entraînant des crises vaso-occlusives et des dommages multi-organiques. Ces globules rouges sont très fragiles, ce qui ajoute une anémie hémolytique au tableau clinique. Dans la β-thalassémie, des centaines de mutations différentes ont été décrites, entraînant une synthèse réduite ou absente de chaîne de globine-β. Cela conduit à la précipitation des chaînes α non couplées en excès, provoquant une érythropoïèse inefficace, l’apoptose des précurseurs érythroïdes, un stress oxydant, la mort des globules rouges et une anémie [3, 4] (Figure 1).
 |
Figure 1. Les β-hemoglobinopathies. L’hémoglobine adulte (HbA), composée de deux chaînes de gioblne-α (gènes localisés sur le chromosome 16) et de deux chaînes de globine-β (gène localisé sur le chromosome 11), constitue le principale composant des globules rouges. Dans la drépanocytose, une mutation ponctuelle A>T (GLU>VAL) provoque la production de la globine-βS, la formation de l’hémoglobine falciforme (HbS) qui polymérise et déforme les globules rouges (globule rouge en faucille), entraînant des crises vaso-occlusives et une anémie. La β-thalassémie est due à >400 mutations réduisant l’expression de la globine-β, et la production d’HbA, et provoquant un excès de chaînes de globine-a non couplées, responsable d’érythropoïèse inefficace et d’anémie. (Créée avec BioRender.com) |
Les traitements typiques des β-hémoglobinopathies sont la transfusion de globules rouges provenant de donneurs sains et les traitements pharmacologiques. Cependant, ces thérapies sont à vie et ne sont pas également efficaces chez tous les patients.
La transplantation de cellules souches/progénitrices hématopoïétiques et la thérapie génique
L’hématopoïèse, le processus de production et de maturation de toutes les cellules sanguines, est assurée par les cellules souches/ progénitrices hématopoïétiques résidant dans la moelle osseuse. La transplantation de cellules souches/progénitrices hématopoïétiques provenant d’un donneur compatible est devenue la norme de soins pour remplacer un système sanguin défectueux par un système sain. Cependant, cette transplantation n’est disponible que pour une fraction de patients ayant un donneur compatible, et les risques associés sont la maladie du greffon contre l’hôte et le rejet du greffon.
La hiérarchie singulière du système hématopoïétique, avec les cellules souches/progénitrices hématopoïétiques à son sommet, se régénérant elles-mêmes et renouvelant les globules rouges et blancs circulants, offre une opportunité unique pour les stratégies de thérapie génique ciblant les maladies du sang, telles que les β-hémoglobinopathies. En particulier, la transplantation de cellules souches/progénitrices hématopoïétiques autologues, génétiquement corrigées ex vivo, représente une option thérapeutique prometteuse pour les patients ne disposant pas de donneur compatible (Figure 2).
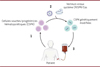 |
Figure 2. La thérapie génique. 1. Les cellules souches/progénltrlces hématopoïétiques (CSPH) sont collectées par aphérèse chez le patient, puis 2. génétiquement modifiées ex vivo dans des laboratoires spécialisés via des vecteurs viraux, porteur de transgène thérapeutique, ou via les technologies de l’édition du génome, telles que le système CRISPR/Cas, pour inactiver des gènes ou des séquences régulatrices ou corriger des gènes défectueux. 3. Les cellules souches/progénitrices hématopoïétiques autologues génétiquement modifiées sont ensuite réinjectées chez le patient. (Créée avec BioRender.com) |
Les premières stratégies de thérapie génique visaient à restaurer la fonction d’un gène perdu et reposaient sur des systèmes viraux recombinants pour délivrer efficacement l’ADN externe dans la cellule cible. En général, l’acide nucléique transféré exprimait une copie fonctionnelle du gène défectueux de l’hôte, une stratégie connue sous le nom de complémentation génique ou d’addition de gènes [5]. Pour modifier toutes les cellules du système hématopoïétique, cette approche nécessite un transfert ex vivo efficace dans les cellules souches/progénitrices hématopoïétiques et l’intégration du transgène thérapeutique. Plus précisément, les cellules souches/progénitrices hématopoïétiques sont prélevées sur les patients, génétiquement modifiées dans des laboratoires spécialisés, puis réinjectées aux patients après qu’ils/elles aient reçu un traitement myéloablatif pour faire de la place dans la moelle osseuse pour les cellules modifiées.
Cependant, l’utilisation de vecteurs intégratifs a été associée à une mutagenèse insertionnelle [6-11]. De plus, les longues séquences régulatrices ne peuvent pas toujours être intégrées dans ces vecteurs, rendant impossible la récapitulation de la régulation naturelle de certains transgènes, ce qui est essentiel pour certains gènes thérapeutiques. Dans le cas des β-hémoglobinopathies, plusieurs essais cliniques ont démontré le bénéfice de l’intégration d’un transgène de globine-β, apporté par un lentivirus [12, 13]. Les niveaux d’expression de ce transgène sont corrélés au nombre de copies de vecteur intégré par cellule. Celui-ci étant variable entre les patients et les études, l’expression du transgène par copie de vecteur demeurait faible par rapport aux niveaux de globine-β endogène, et l’amélioration du phénotype clinique nécessitait l’intégration d’un nombre élevé de vecteurs [14], ce qui ne peut être atteint chez tous les patients et pourrait augmenter le risque de génotoxicité. En outre, la fabrication de vecteurs lentiviraux est très coûteuse. Cette thérapie n’est actuellement commercialisée qu’aux États-Unis, pour un coût de 2,8 à 3,1 millions de dollars par patient.
L’édition du génome par la technologie CRISPR/Cas9
L’édition du génome est aujourd’hui un domaine prolifique générant de nouveaux outils et de nouvelles applications. Cependant, ce n’est que depuis une douzaine d’années que la publication sur les nucléases programmables par l’ARN révolutionne non seulement le domaine de la thérapie génique, mais aussi la modélisation des maladies et la génétique fondamentale.
Au début des années 2010, une découverte révolutionnaire était sur le point de transformer ce domaine. Doudna et Charpentier ont réussi à exploiter l’efficacité de coupe du système hybride ARN-protéine-ADN de l’immunité intracellulaire procaryote pour couper l’ADN [15]. Ce système, appelé CRISPR (clusters of regularly interspaced short palindromic repeats) / Cas9, a été validé comme l’un des outils les plus polyvalents de la biologie moléculaire pour une utilisation dans des cellules eucaryotes [16, 17]. Cela a considérablement réduit les exigences techniques par rapport aux nucléases précédemment utilisées, car le système CRISPR/Cas9 ne nécessite que l’utilisation d’un court ARN (ARN guide, ARNg) capable de se lier simultanément à la protéine Cas9 et au locus ciblé. La nucléase Cas9 possède une activité de coupe spécifique du double brin d’ADN capable de perturber la séquence ciblée [15, 17, 18]. Dans le domaine de la thérapie génique, cette capacité peut être utilisée soit pour inactiver des gènes ou des séquences régulatrices (via le mécanisme de réparation cellulaire par jonction non homologue [NHEJ, non-homologous end joining], qui provoque la formation de petites insertions ou délétions appelées InDels), soit pour corriger une mutation pathologique (via la réparation dirigée par homologie [HDR, homology-directed repair]) grâce à un modèle donneur portant la séquence du gène de type sauvage. Remarquablement, malgré sa découverte récente, cette technologie a déjà été utilisée à des fins diagnostiques [19] et thérapeutiques [20].
Essais cliniques basés sur l’édition du génome des CSPH pour les β-hémoglobinopathies
Malgré le peu de temps écoulé depuis l’adaptation des outils CRISPR à l’édition du génome [15], l’intérêt suscité a été si intense qu’il y a déjà plus de vingt essais cliniques enregistrés dans lesquels des cellules souches/progénitrices hématopoïétiques ont été éditées ex vivo puis réinjectées chez des patients atteints de drépanocytose et de β-thalassémie. Ainsi, les β-hémoglobinopathies dominent actuellement l’application des cellules souches/progénitrices hématopoïétiques génétiquement modifiées.
La majorité de ces essais cliniques vise à réactiver l’expression de l’hémoglobine fœtale (HbF) [21-23]. En effet, la persistance de l’expression des sous-unités de globine-γ (HBG1 et HBG2), composant l’HbF, à l’âge adulte, connue sous le nom de persistance héréditaire de l’hémoglobine fœtale (PHHF) et causée par des variants génétiques bénins, améliore les symptômes des patients atteints de β-hémoglobinopathies. Ces variants génétiques ont incité les scientifiques à étudier les facteurs et les mécanismes de régulation impliqués dans le passage de l’HbF à l’HbA. Les facteurs de transcription, tels que BCL11A, ZBTB7A et MYB, figurent parmi les principaux régulateurs de l’expression de l’HbF. La diminution de l’expression de ces gènes s’est avérée capable de réactiver l’expression de l’HbF [21-26]. Cela a ouvert la voie à l’utilisation d’outils d’édition du génome, tels que le système CRISPR/Cas9, pour inactiver BCL11A (le gène codant un répresseur transcriptionnel majeur de l’HbF), lever la répression de l’HbF et améliorer les symptômes chez ces patients (Figure 3).
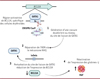 |
Figure 3. Gasgevy® : l’approche CRISPR/Cas9 pour les β-hemoglobinopathies. Dans la thérapie Casgevy®, le système CRISPR/ Cas9 a été façonné pour cibler spécifiquement le site de liaison (SL) de GATAI situé dans une région activatrice du gène BCLIIA. 1. Ce système génère une cassure double brin au niveau de ce site de liaison qui 2. est ensuite réparée via le mécanisme de réparation cellulaire par jonction non homologue (NHEJ, non-homologous end joining), J. provoquant de petites insertions et délétions (InDels), et altérant le site de liaison de GATAI. L’expression de BCL11A est ainsi réduite, supprimant son effet négatif sur les gènes des globines-γ (HBG1 et HBG2). 4. Ces globines sont alors exprimées permettant la production d’hémoglobine fœtale (HbF) dans les cellules érythroïdes provenant des cellules souches/progéni trices hématopoïétiques modifiées. (Créée avec BioRender.com). |
Cette ligne de recherche clinique, qui est la plus avancée, constitue la thérapie commercialisée sous le nom de Casgevy®. Casgevy® (exagamglogene autotemcel) est un produit de thérapie génique développé par Vertex Pharmaceuticals et CRISPR Therapeutics, qui consiste en des cellules souches/progénitrices hématopoïétiques autologues éditées par la technique CRISPR/Cas91. Il s’agit notamment de la première thérapie approuvée utilisant le système CRISPR/ Cas9 comme outil d’édition du génome, seulement dix ans après que les chercheurs aient décrit pour la première fois cette technique (Figure 3) [15]. Casgevy® a été approuvé le 8 décembre 2023 par la FDA (Food and Drug Administration), quelques semaines après son approbation conditionnelle par la MHRA (Medicines and Healthcare products Regulatory Agency)2, pour le traitement des patients âgés de 12 ans et plus atteints de drépanocytose sévère caractérisée par des crises vaso-occlusives récurrentes3. Peu après, la FDA a étendu l’autorisation aux patients atteints de β-thalassémie dépendante des transfusions4 et l’EMA (European Medicines Agency) a accordé une autorisation de mise sur le marché conditionnelle en Europe pour ces deux maladies5. La stratégie Casgevy® est désormais sur le marché américain au prix de 2,2 millions de dollars par patient.
Plus précisément, Casgevy® est préparé à partir des cellules souches/progénitrices hématopoïétiques de chaque patient, qui sont mobilisées dans la circulation sanguine et collectées par aphérèse des cellules du sang périphérique. Les cellules souches/ progénitrices hématopoïétiques CD34+ sont ensuite isolées pour la fabrication du produit thérapeutique, qui consiste à éditer le génome de ces cellules ex vivo, en introduisant le complexe ribonucléoprotéique (RNP) CRISPR/Cas9 par électroporation (Figure 2). L’ARNg du complexe RNP permet à la nucléase Cas9 de produire une cassure double brin précise dans l’ADN. Dans le cadre de Casgevy®, cette cassure double brin de l’ADN est générée au niveau du site de liaison de GATA1 localisé dans la région activatrice, spécifique des cellules érythroïdes, du gène BCL11A (Figure 3). La formation de cette cassure double brin de l’ADN déclenche sa réparation par la voie NHEJ, qui provoque la formation d’InDels, la perturbation de la liaison de l’activateur transcriptionnel GATA1, et par conséquent, la réduction de l’expression de BCL11A (Figure 3). Les cellules souches/progénitrices hématopoïétiques CD34+ modifiées sont ensuite formulées en suspension et réinjectées en une seule dose comme dans le cadre d’une greffe de cellules souches/progénitrices hématopoïétiques, après que le patient ait subi un traitement myéloablatif pour éliminer les cellules souches sanguines malades et faire de la place aux cellules corrigées.
Après administration, les cellules souches/progénitrices hématopoïétiques CD34+ modifiées se greffent dans la moelle osseuse et se différencient vers la lignée érythroïde. Comme BCL11A code un facteur de transcription critique qui réprime l’expression des gènes HBG1 et HBG2, sa répression entraîne une augmentation de la production d’HbF dans les cellules érythroïdes. Chez les patients atteints de drépanocytose sévère, l’HbF réduit les niveaux d’HbS, corrigeant ainsi le phénotype de falciformation des globules rouges et réduisant la fréquence des crises vaso-occlusives et le besoin en transfusions. Chez les patients atteints de β-thalassémie dépendante des transfusions, la production de globine-γ compense le déficit en globine-β, réduisant ainsi l’érythropoïèse inefficace et l’anémie sévère, et élimine la dépendance aux transfusions régulières [25, 27, 28].
Les premiers résultats de l’essai clinique mené par Vertex Pharmaceuticals ont été publiés en 2021 [25]. Le premier patient β-thalassémique et le premier drépanocytaire de cet essai ont présenté des niveaux d’Hb normaux ou proches de la normale grâce à la réactivation de l’HbF. De plus, les niveaux d’HbS ont chuté à environ 50 % chez le patient drépanocytaire (en comparaison des niveaux de 30-40 % d’HbS sont observés chez les porteurs asymptomatiques d’un trait drépanocytaire). Les deux patients ont montré une amélioration substantielle du phénotype clinique (aucune crise vaso-occlusive n’a été observée chez le patient drépanocytaire) et ont atteint l’indépendance transfusionnelle.
Un essai de phase III a été initié pour des patients âgés de 12 à 35 ans atteints de β-thalassémie [28], révélant des taux d’Hb normaux ou proches de la normale (principalement représentés par l’HbF) avec une efficacité d’édition allant de 40 à 85 %. L’indépendance transfusionnelle a été atteinte chez 91 % des patients. Les patients qui n’ont pas atteint l’indépendance transfusionnelle ne correspondaient pas uniquement à des patients avec une faible efficacité d’édition, ce qui suggère que d’autres facteurs liés aux patients eux-mêmes contribuent probablement au succès de cette thérapie. Il est intéressant de noter que deux patients pédiatriques atteints de β-thalassémie ont été traités avec succès lors d’un essai clinique de phase I/II utilisant une stratégie similaire (sponsorisé par Bioray), atteignant des niveaux d’Hb normaux avec une efficacité d’édition de 85-89 % [29].
Un deuxième essai de phase III, sponsorisé par Vertex Pharmaceuticals, pour des patients atteints de drépanocytose âgés de 12 à 35 ans [27] a également confirmé les résultats positifs de cette thérapie avec des niveaux de 50 % d’HbF et de 50 % d’HbS. Sur les 44 patients, 37 n’ont pas présenté de crise vaso-occlusive après le traitement et ont montré une diminution de l’hémolyse et des niveaux d’édition globalement élevés, même chez les patients qui n’ont pas atteint les critères principaux, ce qui suggère une fois de plus que d’autres facteurs peuvent influencer le résultat de ce traitement.
Autres approches d’édition CRISPR/Cas9 pour les β-hémoglobinopathies
D’autres cibles et outils d’édition du génome ont également été testés dans des essais cliniques de phase I/II, tels que cibler les gènes des globines fœtales pour réactiver l’HbF à l’aide d’un autre système de nucléase CRISPR/Cas (CRISPR/AsCas12, Cas12 de Acidibacillus sulfuroxidans) et corriger le gène de la globine-β adulte via le système CRISPR/Cas9 et la voie HDR de réparation de l’ADN.
Plus précisément, l’édition du génome a été exploitée pour corriger le gène endogène de la globine-β en utilisant le système CRISPR/Cas9. Ce système induit une cassure double brin de l’ADN dans le gène de la globine-β, qui est ensuite réparée à l’aide d’une matrice ADN donneuse portant la séquence de la globine-β de type sauvage par la voie HDR [30-32]. Deux essais cliniques fondés sur cette stratégie ont été conçus. Cependant, la voie HDR utilisée dans les approches de correction génique est moins efficace dans les cellules souches/progénitrices hématopoïétiques que l’autre principale voie de réparation de l’ADN, la voie NHEJ. En effet, une efficacité de correction génique de seulement 2,3 % a été observée dans le premier essai clinique sponsorisé par Graphite Bio [33]. De plus, cette stratégie nécessite l’apport d’une matrice donneuse, ce qui peut être complexe et même toxique dans certains cas [32]. Il est à noter que ce même essai, sponsorisé par Graphite Bio, a été volontairement interrompu en raison d’une pancytopénie6 survenue chez le premier patient [33]. Étant donné ces inconvénients des stratégies fondées sur la voie HDR [30, 34], plusieurs approches sont à l’étude pour réduire la toxicité cellulaire et améliorer l’efficacité de l’édition [35-37].
Conclusions et développements futurs
L’approbation de Casgevy® représente une étape importante dans le domaine des thérapies géniques innovantes, soulignant l’énorme potentiel thérapeutique de l’édition du génome pour traiter les causes sous-jacentes des maladies génétiques pour lesquelles il n’existe pas d’options thérapeutiques efficaces. Bien que la sécurité et l’efficacité à long terme de cette approche puissante doivent encore être évaluées, de nouvelles stratégies sont en cours de développement pour améliorer davantage les résultats thérapeutiques, réduire la génotoxicité potentielle et les coûts de la thérapie.
À titre d’exemple, les stratégies basées sur les nucléases génèrent des cassures doubles brins de l’ADN qui peuvent induire une réponse aux dommages de l’ADN, conduisant à l’apoptose et peuvent provoquer d’importants réarrangements génomiques [38]. Par conséquent, l’idée est de développer de plus en plus de stratégies sans cassure double brin de l’ADN pour les β-hémoglobinopathies en utilisant des technologies fondées sur CRISPR/Cas9, telles que les éditeurs de bases (base editors) [39, 40], qui peuvent effectuer les conversions C>T ou A>G, les éditeurs primaires (prime editors) [41], qui peuvent induire toutes les conversions de bases, et les éditeurs de l’épigénome (epigenome editors) [42], qui peuvent activer ou inactiver des gènes en déposant ou supprimant des marques de transcription actives ou inactives, comme les modifications des histones. Par exemple, l’édition des promoteurs des deux gènes des globines-γ fœtales (HBG1 et HBG2) à l’aide de technologies d’édition de bases est actuellement testée dans des essais cliniques de phase I/II [43, 44].
Enfin, l’idée pour l’avenir est de développer une stratégie in vivo fondée sur la mobilisation des cellules souches/progénitrices hématopoïétiques dans la circulation sanguine, où elles deviennent plus accessibles, suivie de l’injection de nanoparticules ou de vecteurs viraux ciblant spécifiquement les cellules souches hématopoïétiques pour délivrer les éditeurs du génome. Cela simplifiera la procédure de thérapie génique, réduira les coûts et la morbidité associés à la chimiothérapie, permettant ainsi une utilisation plus large de ces thérapies.
Remerciements
Ce travail a bénéficié du soutien de l’Agence nationale de la recherche dans le cadre des Investissements d’Avenir (ANR-10-IAHU-01), du Conseil européen de la recherche (865797 DITSB), de la Commission européenne (subvention HORIZON-RIA EDITSCD n° 101057659), et de l’AFM-Téléthon (subventions 22206, 22399 et 23879).
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
STN : 125787 et STN : 125785, www.fda.gov.
STN : 125785, www.fda.gov.
EMEA/H/C/005763, www.ema.europa.eu.
La pancytopénie est un état pathologique caractérisé par une réduction significative du nombre de presque toutes les cellules sanguines (globules rouges, plaquettes, monocytes, lymphocytes, etc.).
Références
- Hardouin G, Magrin E, Corsia A, et al. Sickle Cell Disease: From Genetics to Curative Approaches. Annu Rev Genomics Hum Genet 2023 ; 24 : 255–75. [CrossRef] [PubMed] [Google Scholar]
- Cavazzana M, Corsia A, Brusson M, et al. Treating Sickle Cell Disease: Gene Therapy Approaches. Annu Rev Pharmacol Toxicol 2024 ; 65. doi.org/10.1146/annurev-pharmtox-022124–022000. [PubMed] [Google Scholar]
- Taher AT, Musallam KM, Cappellini MD. β-Thalassemias. N Engl J Med 2021 ; 384 : 727–43. [CrossRef] [PubMed] [Google Scholar]
- Origa R. β-Thalassemia. Genet Med 2017 ; 19 : 609–19. [CrossRef] [PubMed] [Google Scholar]
- Williams DA. The long road traveled in hematopoietic stem cell gene therapy. Mol Ther 2022 ; 30 : 3097–9. [CrossRef] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003 ; 302 : 415–9. [CrossRef] [PubMed] [Google Scholar]
- European Society of Gene Therapy (ESGT). One of three successfully treated CGD patients in a Swiss-German gene therapy trial died due to his underlying disease: A position statement from the European Society of Gene Therapy (ESGT). J Gene Med 2006 ; 8 : 1435. [CrossRef] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Garrigue A, Wang GP, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest 2008 ; 118 : 3132–42. [CrossRef] [PubMed] [Google Scholar]
- Howe SJ, Mansour MR, Schwarzwaelder K, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest 2008 ; 118 : 3143–50. [CrossRef] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 2010 ; 467 : 318–22. [CrossRef] [PubMed] [Google Scholar]
- Cesana D, Cicalese MP, Calabria A, et al. A case of T-cell acute lymphoblastic leukemia in retroviral gene therapy for ADA-SCID. Nat Commun 2024 ; 15 : 3662. [CrossRef] [PubMed] [Google Scholar]
- Locatelli F, Thompson AA, Kwiatkowski JL, et al. Betibeglogene Autotemcel Gene Therapy for Non-β0/β0 Genotype β-Thalassemia. N Engl J Med 2022 ; 386 : 415–27. [CrossRef] [PubMed] [Google Scholar]
- Kanter J, Walters MC, Krishnamurti L, et al. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. N Engl J Med 2022 ; 386 : 617–28. [CrossRef] [PubMed] [Google Scholar]
- Magrin E, Semeraro M, Hebert N, et al. Long-term outcomes of lentiviral gene therapy for the β-hemoglobinopathies: the HGB-205 trial. Nat Med 2022 ; 28 : 81–8. [CrossRef] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012 ; 337 : 816–21. [CrossRef] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013 ; 8 : 2281–308. [CrossRef] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science 2013 ; 339 : 823–6. [CrossRef] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013 ; 339 : 819–23. [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Kellner MJ, Koob JG, Gootenberg JS, et al. SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat Protoc 2019 ; 14 : 2986–3012. [CrossRef] [PubMed] [Google Scholar]
- Paul B, Montoya G. CRISPR-Cas12a: Functional overview and applications. Biomed J 2020 ; 43 : 8–17. [CrossRef] [PubMed] [Google Scholar]
- Bauer DE, Kamran SC, Lessard S, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 2013 ; 342 : 253–7. [CrossRef] [PubMed] [Google Scholar]
- Canver MC, Smith EC, Sher F, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015 ; 527 : 192–7. [CrossRef] [PubMed] [Google Scholar]
- Liu N, Hargreaves VV, Zhu Q, et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 2018 ; 173 : 430–42.e17. [CrossRef] [PubMed] [Google Scholar]
- Steinberg MH. Fetal hemoglobin in sickle cell anemia. Blood 2020 ; 136 : 2392–400. [CrossRef] [PubMed] [Google Scholar]
- Frangoul H, Altshuler D, Cappellini MD, et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N Engl J Med 2021 ; 384 : 252–60. [CrossRef] [PubMed] [Google Scholar]
- Esrick EB, Lehmann LE, Biffi A, et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. N Engl J Med 2021 ; 384 : 205–15. [CrossRef] [PubMed] [Google Scholar]
- Frangoul H, Locatelli F, Sharma A, et al. Exagamglogene Autotemcel for Severe Sickle Cell Disease. N Engl J Med 2024 ; 390 : 1649–62. [CrossRef] [PubMed] [Google Scholar]
- Locatelli F, Lang P, Wall D, et al. Exagamglogene Autotemcel for Transfusion-Dependent β-Thalassemia. N Engl J Med 2024 ; 390 : 1663–76. [CrossRef] [PubMed] [Google Scholar]
- Fu B, Liao J, Chen S, et al. CRISPR-Cas9-mediated gene editing of the BCL11A enhancer for pediatric β0/β0 transfusion-dependent β-thalassemia. Nat Med 2022 ; 28 : 1573–80. [CrossRef] [PubMed] [Google Scholar]
- Lattanzi A, Camarena J, Lahiri P, et al. Development of β-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Sci Transl Med 2021 ; 13 : eabf2444. [CrossRef] [PubMed] [Google Scholar]
- Magis W, DeWitt MA, Wyman SK, et al. High-level correction of the sickle mutation is amplified in vivo during erythroid differentiation. iScience 2022 ; 25 : 104374. [CrossRef] [PubMed] [Google Scholar]
- Moiani A, Letort G, Lizot S, et al. Non-viral DNA delivery and TALEN editing correct the sickle cell mutation in hematopoietic stem cells. Nat Commun 2024 ; 15 : 4965. [CrossRef] [PubMed] [Google Scholar]
- Shyr DC, Lowsky R, Miller W, et al. One Year Follow-up on the First Patient Treated with Nula-Cel: An Autologous CRISPR/Cas9 Gene Corrected CD34+ Cell Product to Treat Sickle Cell Disease. Blood 2023 ; 142 : 5000. [CrossRef] [Google Scholar]
- Uchida N, Li L, Nassehi T, et al. Preclinical evaluation for engraftment of CD34+ cells gene-edited at the sickle cell disease locus in xenograft mouse and non-human primate models. Cell Rep Med 2021 ; 2 : 100247. [CrossRef] [PubMed] [Google Scholar]
- Schiroli G, Conti A, Ferrari S, et al. Precise Gene Editing Preserves Hematopoietic Stem Cell Function following Transient p53-Mediated DNA Damage Response. Cell Stem Cell 2019 ; 24 : 551–65.e8. [CrossRef] [PubMed] [Google Scholar]
- Ferrari S, Jacob A, Beretta S, et al. Efficient gene editing of human long-term hematopoietic stem cells validated by clonal tracking. Nat Biotechnol 2020 ; 38 : 1298–308. [CrossRef] [PubMed] [Google Scholar]
- Selvaraj S, Feist WN, Viel S, et al. High-efficiency transgene integration by homology-directed repair in human primary cells using DNA-PKcs inhibition. Nat Biotechnol 2024 ; 42 : 731–44. [CrossRef] [PubMed] [Google Scholar]
- Amendola M, Brusson M, Miccio A. CRISPRthripsis: The Risk of CRISPR/Cas9-induced Chromothripsis in Gene Therapy. Stem Cells Transl Med 2022 ; 11 : 1003–9. [CrossRef] [PubMed] [Google Scholar]
- Antoniou P, Hardouin G, Martinucci P, et al. Base-editing-mediated dissection of a γ-globin cis-regulatory element for the therapeutic reactivation of fetal hemoglobin expression. Nat Commun 2022 ; 13 : 6618. [CrossRef] [PubMed] [Google Scholar]
- Hardouin G, Antoniou P, Martinucci P, et al. Adenine base editor-mediated correction of the common and severe IVS1-110 (G>A) β-thalassemia mutation. Blood 2023 ; 141 : 1169–79. [CrossRef] [PubMed] [Google Scholar]
- Everette KA, Newby GA, Levine RM, et al. Ex vivo prime editing of patient haematopoietic stem cells rescues sickle-cell disease phenotypes after engraftment in mice. Nat Biomed Eng 2023 ; 7 : 616–28. [CrossRef] [PubMed] [Google Scholar]
- Fontana L, Alahouzou Z, Miccio A, et al. Epigenetic Regulation of β-Globin Genes and the Potential to Treat Hemoglobinopathies through Epigenome Editing. Genes (Basel) 2023 ; 14 : 577. [CrossRef] [PubMed] [Google Scholar]
- Wang L, Lai Y, Liu R, et al. Treatment of patients with severe transfusion-dependent βthalassemia with CS-101, an autologous, ex vivo edited, CD34+ hematopoietic stem cell product using innovative transformer base editor (tHE). Abstract S295 presented at EHA2024 2024. [Google Scholar]
- Han W, Qiu H-Y, Sun S, et al. Base editing of the HBG promoter induces potent fetal hemoglobin expression with no detectable off-target mutations in human HSCs. Cell Stem Cell 2023 ; 30 : 1624–39.e8. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Les β-hemoglobinopathies. L’hémoglobine adulte (HbA), composée de deux chaînes de gioblne-α (gènes localisés sur le chromosome 16) et de deux chaînes de globine-β (gène localisé sur le chromosome 11), constitue le principale composant des globules rouges. Dans la drépanocytose, une mutation ponctuelle A>T (GLU>VAL) provoque la production de la globine-βS, la formation de l’hémoglobine falciforme (HbS) qui polymérise et déforme les globules rouges (globule rouge en faucille), entraînant des crises vaso-occlusives et une anémie. La β-thalassémie est due à >400 mutations réduisant l’expression de la globine-β, et la production d’HbA, et provoquant un excès de chaînes de globine-a non couplées, responsable d’érythropoïèse inefficace et d’anémie. (Créée avec BioRender.com) |
| Dans le texte | |
|
Figure 2. La thérapie génique. 1. Les cellules souches/progénltrlces hématopoïétiques (CSPH) sont collectées par aphérèse chez le patient, puis 2. génétiquement modifiées ex vivo dans des laboratoires spécialisés via des vecteurs viraux, porteur de transgène thérapeutique, ou via les technologies de l’édition du génome, telles que le système CRISPR/Cas, pour inactiver des gènes ou des séquences régulatrices ou corriger des gènes défectueux. 3. Les cellules souches/progénitrices hématopoïétiques autologues génétiquement modifiées sont ensuite réinjectées chez le patient. (Créée avec BioRender.com) |
| Dans le texte | |
|
Figure 3. Gasgevy® : l’approche CRISPR/Cas9 pour les β-hemoglobinopathies. Dans la thérapie Casgevy®, le système CRISPR/ Cas9 a été façonné pour cibler spécifiquement le site de liaison (SL) de GATAI situé dans une région activatrice du gène BCLIIA. 1. Ce système génère une cassure double brin au niveau de ce site de liaison qui 2. est ensuite réparée via le mécanisme de réparation cellulaire par jonction non homologue (NHEJ, non-homologous end joining), J. provoquant de petites insertions et délétions (InDels), et altérant le site de liaison de GATAI. L’expression de BCL11A est ainsi réduite, supprimant son effet négatif sur les gènes des globines-γ (HBG1 et HBG2). 4. Ces globines sont alors exprimées permettant la production d’hémoglobine fœtale (HbF) dans les cellules érythroïdes provenant des cellules souches/progéni trices hématopoïétiques modifiées. (Créée avec BioRender.com). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.