")
")
| Issue |
Med Sci (Paris)
Volume 40, Novembre 2024
Les Cahiers de Myologie
|
|
|---|---|---|
| Page(s) | 60 - 63 | |
| Section | Prix SFM | |
| DOI | https://doi.org/10.1051/medsci/2024133 | |
| Published online | 18 November 2024 | |
Cellules souches musculaires et métabolisme dans la dystrophie musculaire de Duchenne, focus sur la kinase AMPK
Muscle stem cells and metabolism in Duchenne muscular dystrophy, focus on AMPK
Institut NeuroMyoGène, PGNM, CNRS UMR5261/Inserm U1315/ Université Claude Bernard Lyon 1, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Par leur activité myogénique, les cellules souches musculaires (CSM) adultes sont cruciales pour la régénération du muscle strié squelettique. Après activation, elles prolifèrent, se différencient puis fusionnent pour réparer ou former de nouvelles myofibres. Leur progression au cours de la myogenèse nécessite une régulation complexe faisant intervenir plusieurs acteurs tels que le métabolisme, notamment via la kinase AMPK. Cette protéine régule l’auto-renouvellement et l’accrétion myonucléaire des CSM après une lésion aiguë du muscle strié squelettique ou en réponse à la contraction musculaire. Cependant, dans un contexte de dystrophie telle que la myopathie de Duchenne (DMD), la capacité de régénération des CSM est réduite, probablement à cause d’une prolifération accrue au détriment de leur différenciation. Nous nous intéressons ici au potentiel du métabolisme à réguler l’activité myogénique des CSM dans la DMD, par l’intermédiaire de la kinase AMPK.
Abstract
Through their myogenic activity, adult muscle stem cells (MuSCs) are crucial for the regeneration of striated skeletal muscle. Once activated, they proliferate, differentiate and then fuse to repair or form new muscle fibers (myofibers). Their progression through myogenesis requires a complex regulation involving multiple players such as metabolism, in particular via AMPK. This protein kinase regulates the self-renewal and myonuclear accretion of MuSCs after acute skeletal muscle injury or skeletal muscle contraction. However, in a context of dystrophy such as Duchenne muscular dystrophy (DMD), the regenerative capacity of MuSCs is reduced, presumably due to an increase of the proliferation that is detrimental to differentiation. We are interested here in the potential of metabolism to regulate the myogenic activity of MuSCs in DMD via AMPK.
© 2024 médecine/sciences – Inserm
© A. Saugues
Vignette : Myofibre isolée avec deux cellules souches musculaires en division : cellules Pax7 + (vert) et EdU + (blanc).
Représentant 40 % de la masse corporelle, le muscle strié squelettique permet nos mouvements et maintient notre posture grâce à l’activité contractile de ses fibres musculaires (myofibres) [1]. Ces longues cellules sont composées d’une multitude de noyaux post-mitotiques. Leur « maintenance » requiert l’activité de cellules souches musculaires (CSM), en conditions d’homéostasie, de croissance, après un exercice ou une lésion musculaire [1]. Localisées entre le sarcolemme des myofibres et la lame basale, les CSM sont généralement quiescentes. Il s’agit d’un état d’arrêt prolifératif transitoire dont elles peuvent sortir rapidement après détection de changements au sein de leur microenvironnement (ou niche), déclenchés par des stimuli tels que la croissance ou un dommage musculaire. Au cours de la myogenèse, les CSM s’activent, prolifèrent et se différencient en précurseurs musculaires (myocytes), fusionnant soit entre eux pour former une nouvelle fibre musculaire, soit avec une myofibre préexistante, un processus appelé accrétion myonucléaire [2]. En parallèle, une petite portion de CSM activées s’auto-renouvelle, retourne en quiescence pour reconstituer la réserve de CSM [2]. La myogenèse est essentielle à la régénération ainsi qu’à l’homéostasie du muscle strié squelettique.
Ce processus est complexe et dynamique, finement régulé par de multiples facteurs intrinsèques et/ou extrinsèques aux CSM, dont le métabolisme. Effectivement, les CSM sont dotées d’une plasticité métabolique leur permettant d’assurer leurs différents besoins énergétiques au cours de la myogenèse. Les CSM quiescentes utilisent principalement la β-oxydation des acides gras comme source d’énergie, tandis que leur activation requiert une rapide production d’ATP, assurée par la glycolyse. Une reprogrammation métabolique s’effectue lors de leur différenciation, avec un métabolisme plus oxydatif accompagné d’un remodelage du réseau mitochondrial cellulaire [3]. Cette plasticité métabolique est contrôlée par une balance coordonnant les voies cataboliques et anaboliques [4] qui implique le senseur énergétique AMPK (5’-AMP-activated protein kinase), une sérine/thréonine kinase hétérotrimère composée de deux sous-unités régulatrices, β et γ, et d’une sous-unité catalytique a existant sous la forme de deux isoformes [5]. Ce régulateur métabolique s’active en détectant une augmentation des ratios AMP/ATP et ADP/ATP cellulaires, signalant une diminution des niveaux d’énergie. L’AMPK phosphoryle alors ses cibles (protéines et/ou facteurs de transcription) pour réguler plusieurs processus tels que l’absorption du glucose, l’oxydation des acides gras, le métabolisme du glycogène, la synthèse protéique ou encore la biogenèse mitochondriale, pour induire une réponse catabolique et restaurer ainsi les niveaux d’énergie au sein de la cellule [5].
Une étude au sein de notre laboratoire a révélé que les CSM déficientes pour l’AMPKα1 ont un métabolisme principalement glycolytique, augmentant leur taux d’auto-renouvellement et perturbant la régénération musculaire. L’augmentation de la glycolyse est notamment médiée par l’enzyme lactate déshydrogénase (LDH) dont l’activité est accrue en absence de l’AMPKα1 [6]. De plus, nous avons récemment montré que l’isoforme AMPKα2 est essentielle à l’accrétion myonucléaire. Par des approches in vitro et in vivo de traçage cellulaire du devenir des CSM déficientes pour l’AMPKα2, nous avons observé une altération de l’accrétion myonucléaire en réponse à la contraction musculaire, soulignant le rôle de cette isoforme au sein des CSM dans la régulation de ce processus. Dans ce contexte, l’AMPKα2 phosphoryle la protéine BAIAP2 (BAR/IMD domain containing adaptor protein 2), inhibant son rôle négatif dans le remodelage de l’actine, une étape nécessaire pour la fusion entre deux cellules [7].
Ces études ont montré l’importance de l’AMPK dans la régulation des CSM, lorsque leur progression à travers les différentes étapes de la myogenèse est globalement synchrone, après une lésion aiguë du muscle strié squelettique. Cependant, dans le cas de myopathies telles que la dystrophie musculaire de Duchenne (DMD), la régénération musculaire est asynchrone [8], impactant l’activité des CSM.
La dystrophie musculaire de Duchenne (DMD) est une maladie musculaire héréditaire affectant approximativement un garçon sur 3 500 par an. Elle induit une faiblesse musculaire progressive entraînant la perte de la marche, puis l’arrêt respiratoire et/ou cardiaque des patients vers l’âge de 30 ans [9]. Cette pathologie est causée par des mutations au sein du gène DMD, situé sur le chromosome X et codant la dystrophine. Dans le muscle strié squelettique, cette protéine participe à la liaison entre le cytosquelette d’actine et la matrice extracellulaire, permettant la transduction de signaux et la mécanoprotection du sarcolemme. En son absence, ce dernier devient susceptible aux dommages causés par la contraction musculaire. Cela entraîne des cycles asynchrones de dégénérescence/régénération musculaires, engendrant une inflammation chronique [8, 9]. Le muscle dystrophique requiert l’activité des CSM en continu, qui se trouvent alors à coexister à différents stades de la myogenèse, pour tenter de répondre aux différents besoins causés par l’hétérogénéité spatiotemporelle de dégénérescence et de régénération musculaires [8]. Cependant, la régénération musculaire échoue avec le temps. En effet, l’activité myogénique des CSM est perturbée et le tissu musculaire devient graduellement remplacé par de la fibrose et du tissu adipeux [8, 9]. Plusieurs hypothèses pour expliquer cet échec progressif de régénération musculaire chez les patients DMD [8, 9] ont été émises : 1) « l’épuisement » de la réserve des CSM induit par la régénération constante ; 2) la persistance d’un environnement inflammatoire chronique perturbant la niche des CSM ; 3) des défauts intrinsèques aux CSM liés à la déficience en dystrophine ; 4) les signaux contradictoires provenant de microenvironnements de régénération asynchrones. Cette revue fait le point sur les différentes études réalisées pour mieux comprendre l’état des régulations des CSM dans la DMD, avec un focus sur le rôle du métabolisme des CSM dans cette pathologie.
La régulation du devenir des cellules souches musculaires dans la DMD
Il a longtemps été suggéré que le défaut de régénération était dû à l’épuisement du potentiel de régénération des CSM à cause de leur activation répétée et continue dans la DMD [9]. Plusieurs études in vivo ont cependant démontré que le réservoir de CSM était préservé, même à des stades avancés de la pathologie. Que ce soit dans des biopsies de patients DMD ou dans des sections musculaires du modèle murin mdx de la DMD (dont le phénotype est plus léger), le nombre de CSM (exprimant le facteur de transcription PAX7) est similaire, voire plus élevé que dans des muscles sains [10, 11]. Ces études montrent même une activation accrue des CSM, à l’aide de différents marqueurs de prolifération (KI67) et d’activation (MYF5 pour myogenic factor 5). La réserve de CSM persiste donc au fil du temps pour assurer la maintenance du muscle dystrophique. Cependant, ce taux élevé d’activation et de prolifération est suivi d’un défaut de différenciation, avec une réduction du nombre de cellules exprimant le facteur de transcription myogénine dans les muscles de patients DMD [11]. La régénération est donc faible, n’empêchant pas la dégénération et la perte musculaire [11]. Cette capacité de différenciation altérée chez les CSM dystrophiques ne résulterait-elle pas de leur suractivation ? Activées de façon disproportionnée, les CSM pourraient se retrouver bloquées à ce stade, incapables de poursuivre leur progression. Cependant, une étude a montré que l’activation accrue des CSM était une réponse physiologique aux signaux des myofibres. En effet, Anderson et al. montrent que la fragilité sarcolemmale des myofibres chez la souris mdx impacte la signalisation de l’oxyde nitrique connue pour réguler l’activation des CSM [12]. De plus, il semblerait que le défaut de différenciation soit intrinsèque à la cellule, directement causé par la déficience en dystrophine dans les CSM [13]. Dumont et al. ont montré que la dystrophine est exprimée transitoirement au sein des CSM activées et joue un rôle crucial dans la polarité cellulaire, régulant la division asymétrique de ces cellules. En effet, les CSM sont capables de se diviser asymétriquement, pour donner une cellule fille continuant la progression myogénique et une autre cellule fille qui retournera à l’état de quiescence. Lors de cette division, la dystrophine est polarisée avec la protéine MARK2 (microtubule affinity regulating kinase 2) à un pôle de la cellule qui donnera naissance à la cellule fille s’auto-renouvelant. La protéine PARD3 (par-3 family cell polarity regulator) est localisée à l’autre pôle de la cellule qui donnera naissance à la cellule fille progénitrice. Mais en absence de dystrophine, l’alignement apico-basal de l’axe mitotique est altéré. La cellule devient alors incapable de se diviser et entrera en sénescence, réduisant ainsi le nombre de progéniteurs myogéniques participant à la régénération musculaire [13]. Étant donné que les études observent toujours la présence de progéniteurs exprimant la myogénine lors de la progression de la pathologie, bien que réduite selon certaines études [11], d’autres mécanismes semblent donc être impliqués dans la régulation de la différenciation des CSM dans la DMD.
En outre, d’autres études démontrent une différenciation accrue et accélérée des CSM dystrophiques [10, 14]. En effet, deux marqueurs de différenciation (la myogénine et MEF2A pour myocyte enhancer factor 2A) sont exprimés plus rapidement et de façon plus importante chez des CSM mdx in vivo, in vitro et ex vivo, soulignant le caractère autonome à la cellule de cette altération [10, 14]. De plus, un nombre important de myofibres expriment la chaîne lourde de myosine embryonnaire, signe d’une différenciation incomplète dans le muscle dystrophique. Cette expression s’accompagne d’une petite taille des myofibres [10]. La présence de longues chaînes de noyaux centraux marque également l’immaturité persistante de ces cellules [14], signe d’une capacité régénérative des CSM insuffisante et incomplète (Figure 1).
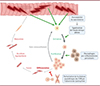 |
Figure 1 Progression altérée des CSM dans la myogenèse dans la DMD. Les lésions musculaires et asynchrones suractivent les CSM, entraînant une accumulation de cellules prolifératives, notamment par l’altération de la signalisation de l’oxyde nitrique et par la présence persistante de macrophages pro-inflammatoires. La déficience en dystrophine impacte la division asymétrique des CSM, réduisant le nombre de progéniteurs myogéniques. La fusion et la maturation des nouvelles fibres formées se retrouvent alors perturbées, résultant en une myogenèse insuffisante. |
Ainsi, la régulation des CSM dans le contexte de la DMD est complexe puisque les résultats sont différents selon le modèle et l’approche utilisés. La dimension spatiotemporelle de la pathologie [8], une variable difficile à mesurer et à prendre en compte, ajoute un niveau de complexité à la compréhension de la physiopathologie de la DMD.
Un rôle de l’AMPK dans la régulation des cellules souches musculaires dystrophiques ?
Comme mentionné plus tôt, l’activation de l’AMPK a des effets pléiotropes, notamment sur la régulation des CSM, au cours de la régénération aiguë et l’homéostasie du muscle squelettique. Dans le cas de la DMD, des études ont montré d’autres rôles de l’AMPK, l’identifiant comme une cible thérapeutique potentielle.
Les fibres rapides qui ont un métabolisme principalement glycolytique sont plus sensibles aux dommages liés aux contractions musculaires dans la DMD que les fibres lentes (ayant un métabolisme plus oxydatif) [15]. La conversion de myofibres via l’activation pharmacologique de l’AMPK dans un modèle dystrophique a permis de rendre le muscle squelettique plus résistant aux dommages grâce à une conversion métabolique oxydative entraînant une augmentation de la proportion de fibres lentes [15]. En plus de son rôle dans la conversion de myofibres dans la DMD, l’activation de l’AMPK a également modulé l’expression de l’utrophine au sein des fibres lentes. Cette protéine homologue à la dystrophine permet ainsi de compenser en partie la perte de la dystrophine liée aux mutations du gène DMD [15]. Cette conversion vers des fibres plus oxydatives pourrait avoir un impact sur les CSM. Un métabolisme plus oxydatif permettrait en effet aux CSM d’entrer en différenciation et de poursuivre la myogenèse [3], plutôt que de rester à un stade d’activation et de prolifération.
Par ailleurs, dans un modèle mdx fibrotique, des macrophages proinflammatoires envahissent en permanence le muscle strié squelettique et sécrètent du TGF-β (transforming growth factor beta), un facteur stimulant la production et la sécrétion du collagène par les fibroblastes, favorisant ainsi la fibrose musculaire [16]. Étant donné que l’AMPK permet la transition des macrophages pro-inflammatoires vers un phénotype réparateur, son activation chez des souris mdx fibrotiques promeut la résolution de l’inflammation en induisant la diminution de facteurs pro-inflammatoires en faveur de facteurs anti-inflammatoires. Juban et al. ont démontré que l’AMPK réduisait l’expression de TGF-β, diminuant ainsi la fibrose, améliorant la régénération musculaire et réduisant le nombre anormalement élevé de CSM dans le muscle [16]. Cette régulation des CSM pourrait s’expliquer par la réduction de facteurs pro-inflammatoires sécrétés par les macrophages, connus pour promouvoir la prolifération des CSM au détriment de leur différenciation [17]. Cette étude démontre ainsi un nouveau rôle de l’AMPK dans la DMD concernant la régulation des macrophages, acteurs communiquant activement avec les CSM et leur microenvironnement.
En conclusion, l’activation de l’AMPK a de multiples effets pléiotropes au sein du muscle squelettique dans le contexte de la DMD. Malgré son implication dans la régulation des CSM, peu d’études ont à ce jour documenté ce rôle dans cette pathologie. De futurs travaux sont donc nécessaires pour valider l’hypothèse de l’implication de l’AMPK dans la régulation des CSM dystrophiques. Bien que l’étude des CSM dans la DMD soit complexe, l’investigation du métabolisme de ces cellules, via notamment l’étude de l’AMPK, permettrait d’apporter des connaissances supplémentaires sur la DMD et, ainsi, de développer de futures pistes thérapeutiques ciblant l’activité des CSM tout en améliorant durablement le phénotype dystrophique.
Prix SFM
Audrey Saugues a reçu le prix Master lors des journées de la Société française de myologie (SFM) 2022.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Frontera WR, Ochala J. Skeletal muscle: a brief review of structure and function. Calcif Tissue Int 2015 ; 96 (3) : 183–195. [CrossRef] [PubMed] [Google Scholar]
- Murach KA, Fry CS, Dupont-Versteegden EE, et al. Fusion and beyond: Satellite cell contributions to loading-induced skeletal muscle adaptation. FASEB J 2021 ; 35 (10) : e21893. [CrossRef] [PubMed] [Google Scholar]
- Relaix F, Bencze M, Borok MJ, et al. Perspectives on skeletal muscle stem cells. Nat Commun 2021 ; 12 (1) : 692. [CrossRef] [PubMed] [Google Scholar]
- Folmes CDL, Dzeja PP, Nelson TJ, et al. Metabolic Plasticity in Stem Cell Homeostasis and Differentiation. Cell Stem Cell 2012 ; 11 (5) : 596–606. [CrossRef] [PubMed] [Google Scholar]
- Mounier R, Théret M, Lantier L, et al. Expanding roles for AMPK in skeletal muscle plasticity. Trends Endocrinol Metab 2015 ; 26 (6) : 275–286. [CrossRef] [PubMed] [Google Scholar]
- Theret M, Gsaier L, Schaffer B, et al. AMPKα1-LDH pathway regulates muscle stem cell self-renewal by controlling metabolic homeostasis. EMBO J 2017 ; 36 (13) : 1946–1962. [CrossRef] [PubMed] [Google Scholar]
- Kneppers A, Ben Larbi S, Theret M, et al. AMPKα2 is a skeletal muscle stem cell intrinsic regulator of myonuclear accretion. iScience 2023 ; 26 (12) : 108343. [CrossRef] [PubMed] [Google Scholar]
- Dadgar S, Wang Z, Johnston H, et al. Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy. J Cell Biol 2014 ; 207 (1) : 139–158. [CrossRef] [PubMed] [Google Scholar]
- Duan D, Goemans N, Takeda S, et al. Duchenne muscular dystrophy. Nat Rev Dis Primer 2021 ; 7 (1) : 13. [CrossRef] [Google Scholar]
- Ribeiro AF, Souza LS, Almeida CF, et al. Muscle satellite cells and impaired late stage regeneration in different murine models for muscular dystrophies. Sci Rep 2019 ; 9 (1) : 11842. [CrossRef] [PubMed] [Google Scholar]
- Kottlors M, Kirschner J. Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res 2010 ; 340 (3) : 541–548. [CrossRef] [PubMed] [Google Scholar]
- Anderson JE. A Role for Nitric Oxide in Muscle Repair: Nitric Oxide-mediated Activation of Muscle Satellite Cells. Mol Biol Cell 2000 ; 11 (5) : 1859–1874. [CrossRef] [PubMed] [Google Scholar]
- Dumont NA, Wang YX, von Maltzahn J, et al. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat Med 2015 ; 21 (12) : 1455–1463. [CrossRef] [PubMed] [Google Scholar]
- Yablonka-Reuveni Z, Anderson JE. Satellite cells from dystrophic (mdx) mice display accelerated differentiation in primary cultures and in isolated myofibers. Dev Dyn Off Publ Am Assoc Anat 2006 ; 235 (1) : 203–212. [Google Scholar]
- Ljubicic V, Khogali S, Renaud JM, et al. Chronic AMPK stimulation attenuates adaptive signaling in dystrophic skeletal muscle. Am J Physiol Cell Physiol 2012 ; 302 (1) : C110–121. [CrossRef] [PubMed] [Google Scholar]
- Juban G, Saclier M, Yacoub-Youssef H, et al. AMPK Activation Regulates LTBP4-Dependent TGF-β1 Secretion by Pro-inflammatory Macrophages and Controls Fibrosis in Duchenne Muscular Dystrophy. Cell Rep 2018 ; 25 (8) : 2163–2176.e6. [CrossRef] [PubMed] [Google Scholar]
- Arnold L, Henry A, Poron F, et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med 2007 ; 204 (5) : 1057–1069. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1 Progression altérée des CSM dans la myogenèse dans la DMD. Les lésions musculaires et asynchrones suractivent les CSM, entraînant une accumulation de cellules prolifératives, notamment par l’altération de la signalisation de l’oxyde nitrique et par la présence persistante de macrophages pro-inflammatoires. La déficience en dystrophine impacte la division asymétrique des CSM, réduisant le nombre de progéniteurs myogéniques. La fusion et la maturation des nouvelles fibres formées se retrouvent alors perturbées, résultant en une myogenèse insuffisante. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.