")
")
| Issue |
Med Sci (Paris)
Volume 40, Number 10, Octobre 2024
|
|
|---|---|---|
| Page(s) | 748 - 756 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2024109 | |
| Published online | 25 October 2024 | |
La correction de mutations pathogènes par Prime editing
Correcting pathogenic mutations using prime editing: an overview
1
Département de médecine moléculaire, Université Laval, Québec, Canada
2
Centre de recherche du CHU de Québec, Université Laval, Québec, Canada
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
L’édition de gènes est un domaine en évolution constante, le Prime editing étant l’une des techniques les plus récentes. Elle permet de modifier un gène sur mesure à l’aide d’une nickase Cas9 qui ne coupe qu’un seul brin d’ADN. Cette nickase est fusionnée à une transcriptase inverse qui recopie en ADN un ARN guide synthétisé à façon. Cette technique est utilisée pour créer des mutations précises dans des modèles cellulaires ou animaux. Le Prime editing est également appliqué en recherche clinique pour traiter des maladies héréditaires, en corrigeant une mutation responsable de l’effet pathogène. Un défi restant est celui de « livrer » un complexe moléculaire thérapeutique aux cellules in vivo. Différentes méthodes sont élaborées pour atteindre les organes propres à chaque maladie.
Abstract
Gene editing is an ever-evolving field and Prime editing technology is among the latest ones. It makes it possible to modify a gene using a Cas9 nickase that cuts a single strand of DNA. This Cas9 nickase is fused with a reverse transcriptase that copies a single guide RNA synthetized by the researcher. This technique is used on one hand to create pathogenic mutations to obtain cell or animal models with a specific mutation. On the other hand, Prime editing is also used in research to treat hereditary diseases by correcting mutations associated with a pathogenic effect. The mode of delivery of the treatment to the affected cells in living organisms constitutes a main challenge. Different methods are studied to reach the organs specific to each disease. This review article presents the latest results in the field as well as the challenges to solve to optimize the possible uses of Prime editing.
Contribution équivalente de premier auteur
© 2024 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (© Wikipedia).
La thérapie génique apporte de grands espoirs dans le traitement des maladies génétiques. Son potentiel réside dans le fait que cette approche touche à l’origine génétique de la maladie, apportant ainsi la possibilité de guérir les maladies héréditaires. Les thérapies géniques ont d’abord eu pour but d’insérer une séquence d’ADN dans le génome pour compenser une mutation récessive [1]. Dans les années 1990-2000, des outils permettant de modifier l’ADN à des sites spécifiques du génome ont été développés, notamment les nucléases à doigt de zinc (ZFN) [2] et les TALEN (transcription activator-like effector nuclease) [3]. La découverte chez la bactérie du système de défense CRISPR/Cas9, puis l’établissement de son mécanisme, en 2012, par Jennifer Doudna et Emmanuelle Charpentier [50] (→), prix Nobel de chimie en 2020, a marqué une étape importante dans le développement des thérapies géniques. Ce système implique une nucléase, la CRISPR associated protein 9 (Cas9), pouvant induire une cassure double brin dans l’ADN. La Cas9 est dirigée vers la séquence à corriger dans le génome par un ARN guide. Ce guide, de 18 à 24 nucléotides (nt), comporte un brin d’ARN complémentaire d’une séquence du génome à modifier [4, 5]. Lorsque le complexe est fixé sur la séquence d’ADN ciblée, la Cas9, si elle reconnaît un motif PAM (protospacer adjacent motif), coupe les deux brins d’ADN, 3 nucléotides en amont de ce motif PAM [6]. Cette séquence PAM varie en fonction du microorganisme d’origine de la Cas9. Par exemple, la Cas9 la plus utilisée, qui provient de Streptococcus pyogenes (SpCas9), reconnaît le PAM 5′-NGG-3′ [7].
(→) Voir le Repères de B. Jordan, m/s n° 1, janvier 2021, page 77
L’évolution et le perfectionnement de la technique CRISPR/Cas9 ont conduit au développement du Base editing (ou édition de base) [8]. Ce système utilise une Cas9 qui contient la mutation D10A (aspartate en position 10 remplacé par une alanine) lui permettant d’induire une coupure d’un seul brin d’ADN, celui contenant le motif PAM [9]. Cette Cas9, endonucléase simple-brin (le terme anglais est nickase), est fusionnée à une cytidine désaminase, ce qui permet de modifier chimiquement, dans une fenêtre de 5 nucléotides, toutes les paires de bases C-G en U-G, et donc remplacées par des paires de bases T-A lors de la réplication. Par la suite, la Cas9 a été fusionnée avec une adénosine désaminase afin de modifier chimiquement les paires de bases A-T et remplacées par la paire de base G-C lors de la réplication. Bien que très pratique, ces deux systèmes ont une application limitée en fonction de la séquence de nucléotides ciblée. En effet, le Base editing ne peut être utilisé que dans les cas où le nucléotide à modifier est unique dans la séquence fenêtre (par exemple, si le nucléotide à modifier est un A, il faut que ce soit le seul A dans une fenêtre de 5 à 10 nucléotides, selon la désaminase utilisée, située entre 14 et 20 nucléotides du motif PAM). Une certaine flexibilité peut être permise dans le cas où les autres nucléotides modifiés dans la fenêtre sont à l’origine de mutations silencieuses ou ne modifient pas l’épissage alternatif de l’ARN pré-messager. Il était donc nécessaire de développer un système plus spécifique et plus souple dans ses capacités de modification.
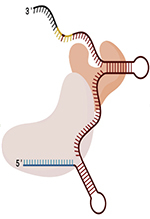
En octobre 2019, le groupe de David R. Liu (Harvard University, ÉtatsUnis) décrit une nouvelle technique, appelée Prime editing [9]. Ce système permet de réaliser des insertions et des délétions ciblées, ainsi que les 12 conversions de bases possibles1. Ce mécanisme permet d’apporter des modifications à l’ADN avec une précision sans précédent et présente des avantages substantiels par rapport au système traditionnel CRISPR/Cas9 et au Base editing (Tableau I). Dérivée du système CRISPR/Cas9, cette nouvelle technique est constituée d’une Cas9 nickase fusionnée à son extrémité carboxyterminale à une transcriptase inverse (RT), et d’un Prime editing guide ARN (pegRNA) (Figure 1). La Cas9 nickase fusionnée à la RT forme ce qui est nommé le Prime editor (PE). Le pegRNA est composé d’une séquence spacer qui s’hybride avec une séquence spécifique de 20 nucléotides d’un brin d’ADN dans le génome. Le pegRNA contient également une séquence primer binding site (PBS) d’environ 10 nucléotides qui s’hybride avec le brin d’ADN coupé par la Cas9 et qui amorcera la transcription inverse. Ce PBS est suivi par un reverse transcriptase template (RTT), séquence modèle d’environ 12 à 35 nucléotides qui contient les modifications qui seront introduites dans le nouveau brin d’ADN synthétisé par la RT. Enfin, le pegRNA contient une région commune structurée, le scaffold, qui se lie à la Cas9 [10] (Figure 1).
 |
Figure 1. Les étapes du Prime editing. Étape 1 : Guidé par la séquence spacer du pegRNA, la Cas9 fusionnée avec une transcriptase inverse (RT) se lie à l’ADN à l’endroit souhaité dans le génome. La Cas9 (ici, la Cas9 de de Streptococcus pyogenes, SpCas9) reconnaît un PAM et induit une coupure simple brin, 3 nucléotides en amont. Étape 2 : Le PBS s’hybride à sa séquence complémentaire sur le brin coupé. Étape 3 : La RT utilise le RTT comme modèle pour rétro transcrire le brin coupé. Étape 4 : Les mésappariements seront réparés par un « 5’ flap » ou un « 3’ flap ». (Figure adaptée de Godbout et al. [11]). pegRNA : Prime editing guide RNA ; PAM : protospacer adjacent motif ; PBS : primer binding site ; RTT : reverse transcriptase template. |
Le processus d’édition du génome débute par la liaison du complexe à l’ADN, le Prime editor étant guidé par la séquence spacer du pegRNA. La Cas9 induit une coupure du brin d’ADN portant le motif PAM, 3 nucléotides en amont de celui-ci. Le PBS s’hybride ensuite à une séquence complémentaire sur le brin coupé de sorte que le pegRNA, qui contient la séquence que l’on souhaite obtenir, serve de modèle pour une transcription inverse. La RT utilise alors le RTT comme modèle pour la synthèse d’un nouveau fragment d’ADN qui prolonge le brin coupé par la Cas9 et contient la mutation souhaitée. Le brin d’ADN prolongé présente donc une section d’ADN proche de celle que l’on souhaite remplacer, de telle sorte qu’il sera nécessaire à la cellule d’éliminer l’une des deux sections pour reconstituer un ADN double brin. Cette zone d’ADN triple brin peut prendre deux configurations : la première avec l’ADN néosynthétisé hybridé au génome cellulaire et le brin original déplacé formant un rabat 5’ (ou 5’ Flap) ; la seconde avec le brin original hybridé à son complémentaire et le brin néosynthétisé formant un rabat 3’ (ou 3’ Flap). Dans le premier cas, si le mésappariement qui résulte de l’introduction de la mutation est réparé en changeant le brin original, et que le 5’ Flap est éliminé, le changement de séquence est réussi. Dans le deuxième cas, si le 3’ Flap est éliminé, la séquence initiale, avant édition, est conservée [9].
Selon la séquence du pegRNA, il est possible d’introduire, des délétions, des insertions (de petite taille) ou des changements d’un seul nucléotide.
Le Prime editing pour corriger ou pour étudier l’effet de mutations pathogènes
Le Prime editing permettant d’effectuer des insertions, des délétions et tous les types possibles de substitutions de nucléotides, cette technique devrait permettre de corriger un important pourcentage de mutations à l’origine de maladies héréditaires. Le Prime editing peut aussi générer des lignées cellulaires ou des modèles animaux présentant des mutations spécifiques, faisant ainsi avancer la recherche sur les maladies génétiques [11]. À ce jour, de nombreuses études précliniques ont montré que cette technique permet de corriger avec précision des mutations dans le génome [11]. Nous présentons quelques-unes des études qui ont utilisé le Prime editing, soit pour créer des modèles d’étude in vitro et in vivo, soit pour traiter des maladies héréditaires, parmi lesquelles des maladies hépatiques, neurologiques, musculaires, ophtalmologiques ou cutanées.
Création de modèles cellulaires ou animaux
Élaboration d’organoïdes et de lignées cellulaires
-
Le déficit en pompe d’export des sels biliaires
ABCB11 (ATP binding cassette subfamily B member 11) code une protéine qui joue le rôle de pompe d’export des sels biliaires dans le foie. Avant de pouvoir corriger la mutation pathogène (D482G, A>G dans le gène ABCB11) à l’origine du déficit en pompe d’exportation des sels biliaires, celle-ci a été introduite dans les cellules d’un organoïde (→) de foie par la technique de Prime editing. Les résultats obtenus par les auteurs de cette étude montrent que la modification se retrouve dans 20 % des cellules de l’organoïde ainsi modifié [12].
-
La mucoviscidose
Geurts et al. [13] ont généré par Prime editing deux organoïdes intestinaux présentant soit la mutation F508del, soit la mutation R785* du gène CFTR (cystic fibrosis transmembrane conductance regulator), deux mutations impliquées dans la mucoviscidose. Bien que les symptômes les plus connus touchent le système respiratoire, les patients atteints de mucoviscidose souffrent également de problèmes intestinaux. En effet, le transport des fluides dans l’intestin dépend de l’activité du canal ionique codé par le gène CFTR. Les organoïdes intestinaux ainsi développés présentent ces mêmes défauts de transport de fluides. Ils constituent donc un modèle intéressant pour étudier la correction génétique des mutations du gène CFTR, la fonction de ce canal pouvant alors être évaluée in vitro.
-
Le déficit immunitaire combiné sévère lié au chromosome X
Hou et al. [14] ont travaillé sur le déficit immunitaire combiné sévère lié à l’X (X-SCID). Cette maladie est causée par des mutations dans le gène codant la sous-unité gamma du récepteur de l’interleukine 2 (IL2RG). Le Prime editing a été utilisé afin de générer un modèle in vitro de la maladie, en introduisant la mutation c.458T>C du gène IL2RG, mutation responsable de la maladie, dans des lymphocytes T de donneurs sains. Utilisant ce modèle, les auteurs ont ensuite pu corriger 26 % des cellules mutées.
-
Les cancers
La mutation G12V du gène KRAS est présente chez certains des patients atteints d’un adénocarcinome pancréatique. Afin d’étudier son effet, cette mutation a été insérée dans le génome de poissons-zèbres. Des mutations off-target (hors cible) ont cependant été observées, en raison d’une séquence homologue à la séquence ciblée par le pegRNA [15]. Chez certains patients atteints de cancer du foie, le gène CTNNB1, qui code la β-caténine, présente une délétion de 6 nucléotides. L’introduction de cette mutation pathogène dans un organoïde de foie a été réalisée par Prime editing avec succès dans 30 % des cellules permettant, là encore, son étude in vitro [16].
-
Le syndrome de Baraitser-Winter et le colobome
Le syndrome cérébrofrontofacial de Baraitser-Winter et le colobome (une malformation congénitale d’une ou plusieurs structures oculaires) sont causés par des mutations du gène ACTB, codant la β-actine. En utilisant la technique du Prime editing, Binder et al. ont réussi à créer des cellules souches pluripotentes présentant la mutation c.640G > T PTC (premature termination codons) dans l’exon 4 du gène ACTB permettant d’examiner les conséquences moléculaires et fonctionnelles du déficit [17].
(→) Voir la série Organoïdes, m/s 2019 à 2024
Création de modèles animaux porteurs de mutations pathogènes
-
Modèle de cataractes
Lin et al. [18] ont généré un modèle de souris souffrant de cataractes en causant une délétion d’un nucléotide (G) dans l’exon 3 du gène CRYGC (crystallin gamma C) par micro-injections de plasmides codant les composants du Prime editing dans des embryons de souris. Cette mutation produit une protéine gamma-cristalline tronquée, ce qui provoque des cataractes nucléaires chez les souris mutantes homozygotes et hétérozygotes. Une souris présentant un taux élevé de délétion du nucléotide (38,2 %) et présentant un phénotype de cataracte nucléaire a ainsi été obtenue. Aucune indel (micro-insertion ou micro-délétion dans la séquence d’ADN) ou mutation hors cible n’ont été détectées. Notons que la mutation, et le phénotype qui lui est associé, sont transmis aux générations suivantes permettant ainsi d’obtenir une lignée de souris malades.
-
Modèle d’albinisme oculocutané
La mutation P302L dans le gène TYR, codant la tyrosinase qui contrôle la synthèse de la mélanine, est responsable de l’albinisme oculocutané. Cette mutation a pu être insérée par la technique de Prime editing dans une lignée de poissons-zèbres avec une fréquence de 3,33 %. Cette étude a par ailleurs montré que la mutation était, comme pour le modèle murin de cataracte, transmise à 8,3 % de la génération suivante [15].
-
Modèle de la maladie de Tay-Sachs
Qian et al. [19] ont généré un modèle de la maladie de Tay-Sachs chez le lapin. Cette maladie neurodégénérative progressive touchant majoritairement la population juive ashkénaze est caractérisée par une accumulation de gangliosides GM2 dans le système nerveux. Elle est due dans 80 % des cas, à une insertion de quatre bases (TATC) dans l’exon 11 du gène HEXA codant l’hexosaminidase A, provoquant un déficit de l’enzyme [20].
-
Modèle d’hypertension pulmonaire
Salem et al. ont inséré, chez la souris, la mutation correspondant à la substitution dans la séquence de la protéine, de la sérine en position 50 par une alanine (Ser50Ala) dans le gène CAPN2 qui code la calpaïne 2, une protéine ubiquitaire ayant des fonctions dans le système nerveux central et dans le cœur. Chez les patients atteints d’hypertension pulmonaire, la sérine en position 50 de la protéine est hyperphosphorylée. Les auteurs ont donc développé ce modèle afin de pouvoir étudier l’impact de la phosphorylation de ce résidu sérine sur la maladie [21].
Le traitement de maladies
In vitro, dans des cellules de patients
-
Le déficit en diacylglycerol O-acyltransferase 1
Le gène DGAT1 code la diacylglycérol O-acyltransférase 1, une enzyme nécessaire pour métaboliser les lipides [12]. Une correction de la mutation S210del (délétion de la sérine en position 210) de la protéine codée par le gène DGAT1, qui cause une entéropathie caractérisée par une diarrhée et une dénutrition chez les nourrissons, a permis d’obtenir 21 % de correction dans des cellules intestinales isolées de patients [22].
-
Les dystrophies rétiniennes liées à CRB1
Les mutations fréquentes du gène CRB1 (crumbs homologue-1), p.(Cys948Tyr)2 et p.(Gly1103Arg), ont été corrigées dans des lignées de cellules souches pluripotentes induites (iPS) de patients avec un taux de réussite de 72 % [23]. Ces mutations peuvent être à l’origine de différents phénotypes, dont l’amaurose congénitale de Leber [24] et la rétinite pigmentaire, qui conduisent à une perte de la vision.
-
La dystrophie musculaire de Duchenne
La dystrophie musculaire de Duchenne est causée par des mutations du gène DMD, codant la dystrophine. Ces mutations sont soit des délétions d’un ou plusieurs exons, qui modifient le cadre de lecture, soit des mutations ponctuelles créant la plupart du temps des codons nonsens. La dystrophine lie la membrane plasmique des fibres musculaires au cytosquelette lors de la contraction. Mbakam et al. ont corrigé la mutation c.428 G>A3 (avec un taux de réussite de 28 %) après 3 traitements par électroporation, et la mutation c.8713C>T (à 22 %), dans des myoblastes de patients en culture [25, 26]. Ces auteurs ont également testés l’insertion de mutations silencieuses et différentes combinaisons de tailles des séquences RTT et PBS. Par ailleurs, Zhao et al. ont réussi à corriger la mutation c.7893delC du gène DMD dans des fibroblastes de patients avec un taux de réussite de 31 % [27].
-
Les maladies liées au gène RYR1
Les maladies liées au gène RYR1 sont causées par une défaillance du canal calcium RyR1 dans les fibres musculaires squelettiques, étant à l’origine chez les patients d’un large spectre phénotypique, allant de la fatigue musculaire, aux crampes, en passant par le syndrome d’hyperthermie maligne. Godbout et al. [28] ont généré par Prime editing, à partir de myoblastes normaux, une lignée de myoblastes présentant la mutation p.(Thr4709Met) dans le gène RYR1 à l’état homozygote. En livrant les composants du Prime editing sous la forme d’ARN, ces auteurs ont ensuite réussi à corriger la mutation avec un taux de réussite de 59 % après 3 traitements de la lignée de myoblastes mutés.
-
L’anémie de Fanconi
Comme plus de 400 autres, la mutation c.1A>G4 du gène FANCA est responsable de l’anémie de Fanconi. Cette maladie se traduit par un déficit, au niveau de la moelle osseuse, de production d’hématies, de leukocytes et de plaquettes sanguines, causant des malformations congénitales, du visage et des mains, et augmentant le risque de certains cancers (leucémie, cancers de la bouche, du cou, de l’anus, ou de la vulve). Le Prime editing permet la correction de la mutation c.1A>G du gène FANCA avec un taux de réussite de 15 % dans des fibroblastes de patients [29].
-
La granulomatose septique chronique
La granulomatose septique chronique est causée par des mutations du gène NCF1 (neutrophil cytosol factor 1), codant la protéine p47phox, une sous-unité de la NADPH oxydase. Cette maladie altère le fonctionnement des neutrophiles, rendant les patients plus susceptibles aux d’infections. La majorité des patients présentent une délétion homozygote de 2 nucléotides (delGT) dans l’exon 2 du gène. Heath et al. [30] ont réussi à corriger au moins un allèle dans 75 % des cellules myéloïdes CD34+ portant la délétion delGT.
-
Les thalassémies alpha
L’une des thalassémies alpha est souvent causée par la mutation CD142 (UAA >CAA) dans le gène HBA2 codant la sous-unité alpha 2 de l’hémoglobine. Shao et al. ont utilisé le Prime editing pour remédier à cette anémie et ont réussi à corriger 39 % des cellules HUDEP2 (human umbilical cord-derived erythroid progenitor) Hb CS (hemoglobin constant spring) traitées [31].
-
L’hypogonadisme primaire héréditaire
L’hypogonadisme causé par la mutation W495X du gène LHCGR (luteinizing hormone/choriogonadotropin receptor), a pour résultat une production insuffisante de testostérone et de spermatozoïdes. Par Prime editing, Xia et al. [32] ont réussi la correction de 23,42 % des cellules de Leydig5 de souris mutées LhcgrW495X, en délivrant tous les éléments du Prime editing à l’aide de lentivirus.
-
Le syndrome de Lesch-Nyhan
Des mutations du gène HPRT1 (hypoxanthine phosphoribosyltransferase 1) sont la cause du syndrome de Lesch-Nyhan, une maladie caractérisée par une hypotonie et un retard développemental. Par Prime editing, Jang et al. [33] ont corrigé jusqu’à 14 % de la mutation c.333_334ins(A) dans des fibroblastes de patients, sans observer de mutations hors cible (off-target).
In vivo, dans des modèles murins
-
L’épilepsie
Dans un modèle de souris portant la mutation R207W (c.619C> T) du gène Kcnq2 (potassium voltage-gated channel subfamily Q member 2), responsable de l’épilepsie, Cao et al. [34] ont utilisé le Prime editing et ont corrigé 14 % des allèles mutés.
-
Le déficit en alpha-1-antitrypsine
La mutation du gène SERPINA1 (serpin peptidase inhibitor family A member 1) cause un déficit en alpha-1-antitrypsine. Cette protéine, produite par le foie, régule l’activité de plusieurs enzymes et a un rôle protecteur dans les poumons. Un déficit en cette protéine cause des dommages aux poumons, au foie et est à l’origine d’une inflammation cutanée [35–37]. La mutation E342K G>A du gène SERPINA1 (allèle PiZ) a été corrigée à 6,7 % dans des souris par injection hydrodynamique intraveineuse d’ADN plasmidique codant les différents composants du Prime editing [37].
-
La phénylcétonurie
Böck et al. [38] ont étudié la correction par Prime editing de la mutation F263S (T>C) du gène Pahenu2 causant la phénylcétonurie, une maladie qui altère la dégradation de la phénylalanine apportée par l’alimentation. En livrant les composants du Prime editing par un adénovirus dans des souriceaux nouveau-nés, ces auteurs sont parvenus à une modification du gène de 11,1 %. Chez les patients, le variant c.1222C>T (p.Arg408Trp) du gène est le plus fréquent. Il a été corrigé en moyenne à 41 % dans le foie de souris p.Arg408Trp par Brooks et al. [39], les composants du Prime editing ayant été livrés par deux vecteurs AAV8 (dual-AAV [adeno-associated virus] de sérotype 8).
-
La tyrosinémie de type I
La tyrosinémie de type I (HT-1) est une maladie touchant le catabolisme de la tyrosine, due à un déficit de l’enzyme fumarylacétoacétate hydrolase (FAH). Chez la souris, elle a pour origine la mutation c.706G>A dans le gène FAH. Jang et al. [24] ont réussi à corriger le déficit in vivo avec un taux de réussite de 11,5 %. La correction a été réalisée en injectant par voie intra-veineuse des plasmides codant les composants du Prime editing. Après traitement, 61 % des hépatocytes exprimaient le gène FAH corrigé. Ce pourcentage est supérieur au taux d’édition de l’ADN génomique, car la plupart des hépatocytes sont polyploïdes [40]. La présence d’ADN provenant d’autres cellules peut également expliquer cette différence [24, 41, 42]. Aucune mutation non désirée n’a été détectée par les auteurs.
-
La microphtalmie et la rétinite pigmentaire
Les souris rd6 représentent un modèle de dégénérescence rétinienne récessive causée par une délétion de 4 pb dans le gène Mfrp (membrane frizzled-related protein) dans un site de donneur d’épissage. Chez l’homme, un défaut génétique dans ce gène induit microphtalmie et rétinite pigmentaire. Le groupe de David Liu [43] a réussi la correction de cette mutation avec un taux de réussite de 15 % dans les yeux des souris rd6. L’injection sous-rétinienne de particules pseudovirales (ou virus-like particles, VLP)6 a permis de délivrer de façon transitoire l’ensemble des composants du Prime editing. La correction de 15 % obtenue a permis de rétablir l’expression de la protéine MFRP dans les yeux traités. Aucune mutation hors cible n’a été détectée, et aucun signe de toxicité ou de modification de la morphologie n’a été observé dans les cryosections des yeux traités.
-
L’amaurose congénitale de Leber
Le groupe de David Liu [43] a également corrigé, par Prime editing délivré par injection sous-rétinienne de particules pseudovirales, la mutation R44X (c.130C>T) du gène RPE65 (retinal pigment epithelium) chez la souris rd12, un modèle de dégénérescence rétinienne. Une correction moyenne de 7,2 % de l’épithélium pigmentaire rétinien a été obtenue. Les souris traitées ont montré une amélioration significative de leur fonction visuelle évaluée par électro-rétinographie. Aucune mutation hors-cible n’a été détectée.
-
L’épidermolyse bulleuse dystrophique récessive
Hong et al. [44] ont corrigé les mutations c.3631C>T et c.2005C>T du gène COL7A1 (collagen type VII alpha 1 chain) avec des taux respectifs de 10,5 et 5,2 %. Ces mutations entraînent une perte de fonction de la protéine de collagène de type VII (C7), provoquant une fragilité de la peau, à l’origine de l’épidermolyse bulleuse dystrophique récessive (RDEB). Le traitement par Prime editing a été réalisé ex vivo sur des fibroblastes dérivés de patients puis transplantés dans des souris nude athymiques immunodéficientes. Dans la peau des souris ainsi traitées, le collagène a pu être restauré à 49 %, un niveau supérieur à celui de 35 % requis pour assurer la stabilité mécanique de la peau [45] et apporter une amélioration thérapeutique. Les fibroblastes corrigés présentent une prolifération accrue par rapport aux cellules non modifiées, expliquant pourquoi le niveaux de restauration du collagène est plus élevé que celui théoriquement attendu en fonction de la fréquence d’édition.
-
La drépanocytose
La drépanocytose est causée par une mutation unique et ponctuelle dans le gène HBB codant la sous-unité β de l’hémoglobine. La mutation consiste dans le changement d’un acide glutamique en une valine. En faible pO2, les tétramères d’hémoglobine s’agrègent entraînant la déformation en faucille des hématies. Le Prime editing a été utilisé ex-vivo sur les cellules souches hématopoïétiques de patients et a permis de corriger entre 15 % et 41 % des cellules. Ces cellules ont été greffées à des souris qui, 17 semaines après la greffe, présentaient entre 28 % et 43 % d’hémoglobine fonctionnelle [46].
-
L’hypercholestérolémie
Chen et al. [47] ont atteint 13 % de correction du gène PCSK9 qui code la pro-protéine convertase subtilisine/kexine de type 9, une enzyme qui oriente vers la dégradation les récepteurs des lipoprotéines de basse densité (LDLR). Le traitement a été réalisé par injection intra-veineuse rétro-orbitale de nanoparticules lipidiques (LNP) contenant les composants du Prime editing chez des souris immunodéficientes. Les auteurs visaient l’insertion de 4 pb (TTAC) dans le gène PCSK9 dans le but de décaler le cadre de lecture afin d’introduire un codon de terminaison prématuré et ainsi inactiver le gène. Inactiver le gène PCSK9 est une stratégie de choix dans le traitement de l’hypercholestérolémie, car elle permet de réduire les concentrations sanguines de LDL-cholestérol (LDL-C) sans induire d’effets délétères.
Les limites du Prime editing
La principale limite du Prime editing reste le mode de délivrance in vivo. En raison de la grande taille du Prime editor, les systèmes qui sont utilisés pour délivrer les composantes de CRISPR-Cas9 ou du Base editing ne fonctionnent pas de façon optimale dans leur forme actuelle pour les composantes du Prime editing. Le développement de nouveaux modes de livraison, ou l’optimisation de vecteurs déjà développés, est donc nécessaire pour une application clinique du Prime editing.
In vivo, plusieurs méthodes ont été expérimentées chez la souris : les micro-injections (dans les embryons) [18] ; les injections hydrodynamiques, dans la veine de la queue [24, 37] ; les injections sous-rétiniennes d’AAV2 (pour le ciblage des yeux) [43], d’AAV8 [39] et d’adénovirus (AdV) [38]. En ce qui concerne les injections, elles ne peuvent être réalisées que dans les organes qui peuvent être ciblés par une intervention peu ou pas invasive, comme les yeux avec des injections sous-rétiniennes [43].
L’expression par les AAV nécessite l’utilisation d’un Prime editor scindé en plusieurs fragments. Cette option n’est cependant pas à privilégier car elle diminue considérablement l’efficacité du Prime editing. L’optimisation de l’assemblage des portions N-et C-terminales du Prime editor est étudiée pour améliorer l’efficacité d’une livraison par double AAV [48]. L’utilisation d’un vecteur adénoviral pour délivrer les composants du Prime editing n’est pas envisageable in vivo, car ce type de vecteur déclenche une importante réaction immunitaire, le développement des vecteurs gutless serait une solution.
Les nanoparticules lipidiques ont également été testées in vivo pour délivrer par voie intra-veineuse le Prime editing à des hépatocytes chez la souris, mais leur efficacité reste très limitée [47]. Les pseudoparticules virales (VLP) sont également des candidats potentiels. Dans des modèles murins, l’efficacité d’une livraison par des VLP au cerveau (par injection intra-cérébro-ventriculaire) et aux yeux (par injection sous-rétinienne) a été démontrée [43]. Dans ce cas, l’efficacité de livraison par les VLP est comparable à celle des AAV, mais avec l’avantage d’utiliser un système de production transitoire des composants du Prime editing.
Dans les traitements ex vivo, le Prime editing est administré directement à des cellules isolées et mises en culture. Il n’est donc pas obligatoire d’utiliser un vecteur viral pour atteindre les cellules à modifier, contrairement au traitement réalisé in vivo. Leur transplantation permet de fournir aux animaux déficients des cellules qui ont été génétiquement corrigées ex vivo [44]. Celle-ci est très efficace dans le cas du foie : les cellules corrigées possèdent en effet un meilleur potentiel de prolifération ; elles peuvent donc repeupler le foie et le rendre à nouveau fonctionnel. Ilest donc plus avantageux de cibler un organe dans lequel les cellules transplantées pourront remplacer les cellules déficientes. La greffe de cellules souches est une autre approche développée par plusieurs équipes [49].
Perspectives
Comme le montre cette revue de la littérature, qui relate l’état des connaissances et des résultats obtenus en corrigeant des mutations par Prime editing, depuis la première publication en octobre 2019 jusqu’à aujourd’hui (début 2024), le Prime editing présente un potentiel certain pour le traitement de maladies génétiques. Technique très récente, des ajustements restent nécessaires pour améliorer son efficacité de modification génomique et sa sécurité vis-à-vis des coupures hors cible, en vue d’une éventuelle application clinique. En particulier, des études sur l’absence de mutations hors cible doivent être menées à l’échelle du génome pour prouver son innocuité. La correction de mutations récessives pourrait être atteinte rapidement. Restaurer un phénotype sain dans ce cas, nécessite théoriquement entre 25 et 75 % de correction, un taux que plusieurs études ont atteint ou sont près d’atteindre, notamment in vitro. Pour la correction de mutations dominantes, une plus grande efficacité d’édition sera nécessaire, et dans plusieurs cas, déterminer le pourcentage de correction permettant une amélioration phénotypique sera important.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Chacune des bases A, T, C ou G qui composent notre ADN peut être remplacée par l’une des trois autres possibilités.
p.(Cys948Tyr) : p. pour protéine, Cystéine en position 948 de la séquence protéique substituée en Tyrosine. p.(Gly1103Arg) : Glycine en position 1 103 substituée en Arginine.
c.428 G>A : c. pour séquence codante, Guanine 428 substituée en Adénine. c.7893delC : délétion de la Cytosine 7893 de la séquence codante.
c.1A>G : Substitution de l’adénine en position 1 en guanine.
Cellule située dans le tissu interstitiel testiculaire, qui élabore la testostérone et forme des petits amas au contact des vaisseaux, entre les tubes séminifères.
Ces pseudo-particules virales sont des particules sans génome viral (pseudo-virions) obtenues par l’assemblage de protéines d’un rétrovirus en présence de formes fusionnées à la nickase d’une partie de ces protéines de structure du virus. Les particules pseudo-virales ne contiennent aucun matériel génétique autre que le pegRNA recruté par introduction d’une séquence de phage.
Références
- Tamura R, Toda M. Historic Overview of Genetic Engineering Technologies for Human Gene Therapy. Neurol Med Chir (Tokyo) 2020 ; 60 : 483–91. [CrossRef] [PubMed] [Google Scholar]
- Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet 2010 ; 11 : 636–46. [CrossRef] [PubMed] [Google Scholar]
- Sun N, Zhao H. Transcription activator-like effector nucleases (TALENs): a highly efficient and versatile tool for genome editing. Biotechnol Bioeng 2013 ; 110 : 1811–21. [CrossRef] [PubMed] [Google Scholar]
- Zhang F, Wen Y, Guo X. CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum Mol Genet 2014 ; 23 : R40–6. [CrossRef] [PubMed] [Google Scholar]
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 2014 ; 32 : 347–55. [CrossRef] [PubMed] [Google Scholar]
- Wu X, Kriz AJ, Sharp PA. Target specificity of the CRISPR-Cas9 system. Quant Biol 2014 ; 2 : 59–70. [CrossRef] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012 ; 337 : 816–21. [CrossRef] [PubMed] [Google Scholar]
- Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017 ; 551 : 464–71. [CrossRef] [PubMed] [Google Scholar]
- Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019 ; 576 : 149–57. [CrossRef] [PubMed] [Google Scholar]
- Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 2020 ; 38 : 824–44. [CrossRef] [PubMed] [Google Scholar]
- Godbout K, Tremblay JP. Prime Editing for Human Gene Therapy: Where Are We Now? Cells 2023 ; 12 : 536. [CrossRef] [PubMed] [Google Scholar]
- Schene IF, Joore IP, Oka R, et al. Prime editing for functional repair in patient-derived disease models. Nat Commun 2020 ; 11 : 5352. [CrossRef] [PubMed] [Google Scholar]
- Geurts MH, de Poel E, Pleguezuelos-Manzano C, et al. Evaluating CRISPR-based prime editing for cancer modeling and CFTR repair in organoids. Life Sci Alliance 2021 ; 4 : e202000940. [CrossRef] [PubMed] [Google Scholar]
- Hou Y, Ureña-Bailén G, Mohammadian Gol T, et al. Challenges in Gene Therapy for Somatic Reverted Mosaicism in X-Linked Combined Immunodeficiency by CRISPR/Cas9 and Prime Editing. Genes (Basel) 2022 ; 13 : 2348. [CrossRef] [PubMed] [Google Scholar]
- Petri K, Zhang W, Ma J, et al. CRISPR prime editing with ribonucleoprotein complexes in zebrafish and primary human cells. Nat Biotechnol 2022 ; 40 : 189–93. [CrossRef] [PubMed] [Google Scholar]
- Abuhamad AY, Mohamad Zamberi NN, Sheen L, et al. Reverting TP53 Mutation in Breast Cancer Cells: Prime Editing Workflow and Technical Considerations. Cells 2022 ; 11 : 1612. [CrossRef] [PubMed] [Google Scholar]
- Binder S, Ramachandran H, Hildebrandt B, Dobner J, Rossi A. Prime-Editing of human ACTB in induced pluripotent stem cells to model human ACTB Loss-of-Function diseases and compensatory mechanisms. Stem Cell Res 2024 ; 75 : 103304. [CrossRef] [PubMed] [Google Scholar]
- Lin J, Liu X, Lu Z, et al. Modeling a cataract disorder in mice with prime editing. Mol Ther Nucleic Acids 2021 ; 25 : 494–501. [CrossRef] [PubMed] [Google Scholar]
- Qian Y, Zhao D, Sui T, et al. Efficient and precise generation of Tay-Sachs disease model in rabbit by prime editing system. Cell Discov 2021 ; 7 : 50. [CrossRef] [PubMed] [Google Scholar]
- Frisch A, Colombo R, Michaelovsky E, et al. Origin and spread of the 1278 insTATC mutation causing Tay-Sachs disease in Ashkenazi Jews: genetic drift as a robust and parsimonious hypothesis. Hum Genet 2004 ; 114 : 366–76. [CrossRef] [PubMed] [Google Scholar]
- Salem AR, Bryant WB, Doja J, et al. Prime editing in mice with an engineered pegRNA. Vascul Pharmacol 2024 ; 154 : 107269. [CrossRef] [PubMed] [Google Scholar]
- Bijker LE, Uyttebroeck S, Hauser B, Vandenplas Y, Huysentruyt K. Variants in DGAT1 causing enteropathy: a case report and review of the literature. Belgian Journal of Paediatrics 2021 ; 23 : 275–9. [Google Scholar]
- Lopes Costa B, Sousa AA, Tsai YT, et al. Development of a Prime Editing Strategy to Treat Mutations in the Crumbs Homologue 1 (CRB1) Gene. Investigative Ophthalmology & Visual Science 2023 ; 64 : 3862. [Google Scholar]
- Jang H, Jo DH, Cho CS, et al. Application of prime editing to the correction of mutations and phenotypes in adult mice with liver and eye diseases. Nat Biomed Eng 2022 ; 6 : 181–94. [Google Scholar]
- Happi Mbakam C, Rousseau J, Lu Y, et al. Prime editing optimized RTT permits the correction of the c.8713C>T mutation in DMD gene. Mol Ther Nucleic Acids 2022 ; 30 : 272–85. [CrossRef] [PubMed] [Google Scholar]
- Happi Mbakam C, Rousseau J, Tremblay G, Yameogo P, Tremblay JP. Prime Editing Permits the Introduction of Specific Mutations in the Gene Responsible for Duchenne Muscular Dystrophy. Int J Mol Sci 2022 ; 23 : 6160. [CrossRef] [PubMed] [Google Scholar]
- Zhao X, Qu K, Curci B, et al. Comparison of In-Frame Deletion, Homology-Directed Repair, and Prime Editing-Based Correction of Duchenne Muscular Dystrophy Mutations. Biomolecules 2023 ; 13 : 870. [CrossRef] [PubMed] [Google Scholar]
- Godbout K, Rousseau J, Tremblay JP. Successful Correction by Prime Editing of a Mutation in the RYR1 Gene Responsible for a Myopathy. Cells 2023 ; 13 : 31. [CrossRef] [PubMed] [Google Scholar]
- Peterkova L, Racková M, Svaton M, et al. P754: prime editing as a novel tool for precise correction of causal mutations in fanconi anaemia group a patient-derived cells. Hemasphere 2023 ; 7 : e27248b5. [CrossRef] [Google Scholar]
- Heath JM, Orenstein JS, Tedeschi JG, et al. Prime Editing Efficiently and Precisely Corrects Causative Mutation in Chronic Granulomatous Disease, Restoring Myeloid Function: Toward Development of a Prime Edited Autologous Hematopoietic Stem Cell Therapy. Blood 2023 ; 142 : 7129. [CrossRef] [Google Scholar]
- Shao C, Liu Q, Xu J, et al. Prime Editing of the a-Thalassemia Hb Constant Spring Mutation. Blood 2023 ; 142 : 5001. [CrossRef] [Google Scholar]
- Xia K, Wang F, Tan Z, et al. Precise Correction of Lhcgr Mutation in Stem Leydig Cells by Prime Editing Rescues Hereditary Primary Hypogonadism in Mice. Adv Sci (Weinh) 2023 ; 10 : e2300993. [CrossRef] [PubMed] [Google Scholar]
- Jang G, Shin HR, Do HS, et al. Therapeutic gene correction for Lesch-Nyhan syndrome using CRISPR-mediated base and prime editing. Mol Ther Nucleic Acids 2023 ; 31 : 586–95. [CrossRef] [PubMed] [Google Scholar]
- Cao BR, Huang YM, Tian FY, et al. Prime editing-based gene correction alleviates the hyperexcitable phenotype and seizures of a genetic epilepsy mouse model. Acta Pharmacol Sin 2023 ; 44 : 2342–5. [CrossRef] [PubMed] [Google Scholar]
- de Serres FJ, Blanco I, Fernández-Bustillo E. Health implications of alpha1-antitrypsin deficiency in Sub-Sahara African countries and their emigrants in Europe and the New World. Genet Med 2005 ; 7 : 175–84. [CrossRef] [PubMed] [Google Scholar]
- Habib O, Habib G, Hwang GH, Bae S. Comprehensive analysis of prime editing outcomes in human embryonic stem cells. Nucleic Acids Res 2022 ; 50 : 1187–97. [CrossRef] [PubMed] [Google Scholar]
- Liu P, Liang SQ, Zheng C, et al. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat Commun 2021 ; 12 : 2121. [CrossRef] [PubMed] [Google Scholar]
- Böck D, Rothgangl T, Villiger L, et al. In vivo prime editing of a metabolic liver disease in mice. Sci Transl Med 2022 ; 14 : eabl9238. [CrossRef] [PubMed] [Google Scholar]
- Brooks DL, Whittaker MN, Qu P, et al. Efficient in vivo prime editing corrects the most frequent phenylketonuria variant, associated with high unmet medical need. Am J Hum Genet 2023 ; 110 : 2003–14. [CrossRef] [PubMed] [Google Scholar]
- Wilkinson PD, Delgado ER, Alencastro F, et al. The Polyploid State Restricts Hepatocyte Proliferation and Liver Regeneration in Mice. Hepatology 2019 ; 69 : 1242–58. [CrossRef] [PubMed] [Google Scholar]
- Kim Y, Hong SA, Yu J, et al. Adenine base editing and prime editing of chemically derived hepatic progenitors rescue genetic liver disease. Cell Stem Cell 2021 ; 28 : 1614–24.e5. [CrossRef] [PubMed] [Google Scholar]
- Jiang T, Zhang XO, Weng Z, Xue W. Deletion and replacement of long genomic sequences using prime editing. Nat Biotechnol 2022 ; 40 : 227–34. [CrossRef] [PubMed] [Google Scholar]
- An M, Raguram A, Du SW, et al. Engineered virus-like particles for transient delivery of prime editor ribonucleoprotein complexes in vivo. Nat Biotechnol 2024 : 10.1038/s41587-023-02078-y. [PubMed] [Google Scholar]
- Hong SA, Kim SE, Lee AY, et al. Therapeutic base editing and prime editing of COL7A1 mutations in recessive dystrophic epidermolysis bullosa. Mol Ther 2022 ; 30 : 2664–2679. [CrossRef] [PubMed] [Google Scholar]
- Kern JS, Loeckermann S, Fritsch A, et al. Mechanisms of fibroblast cell therapy for dystrophic epidermolysis bullosa: high stability of collagen VII favors long-term skin integrity. Mol Ther 2009 ; 17 : 1605–15. [CrossRef] [PubMed] [Google Scholar]
- Everette KA, Newby GA, Levine RM, et al. Ex vivo prime editing of patient haematopoietic stem cells rescues sickle-cell disease phenotypes after engraftment in mice. Nat Biomed Eng 2023 ; 7 : 616–28. [CrossRef] [PubMed] [Google Scholar]
- Chen Z, Kelly K, Cheng H, et al. In Vivo Prime Editing by Lipid Nanoparticle Co-Delivery of Chemically Modified pegRNA and Prime Editor mRNA. GEN Biotechnology 2023 ; 2 : 490–502. [CrossRef] [Google Scholar]
- Davis JR, Banskota S, Levy JM, et al. Efficient prime editing in mouse brain, liver and heart with dual AAVs. Nat Biotechnol 2024 Feb;42(2):253–64. [CrossRef] [PubMed] [Google Scholar]
- Li M, Zhong A, Wu Y, et al. Transient inhibition of p53 enhances prime editing and cytosine base-editing efficiencies in human pluripotent stem cells. Nat Commun 2022 ; 13 : 6354. [CrossRef] [PubMed] [Google Scholar]
- Jordan B. CRISPR : le Nobel, enfin… Med Sci (Paris) 2021 ; 37 : 77–80. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des tableaux
Les avantages et les inconvénients du système CRISPR/Cas9, du Base editing et du Prime editing.1 Possibilité de déterminer la finalité de l’édition.
Liste des figures
|
Figure 1. Les étapes du Prime editing. Étape 1 : Guidé par la séquence spacer du pegRNA, la Cas9 fusionnée avec une transcriptase inverse (RT) se lie à l’ADN à l’endroit souhaité dans le génome. La Cas9 (ici, la Cas9 de de Streptococcus pyogenes, SpCas9) reconnaît un PAM et induit une coupure simple brin, 3 nucléotides en amont. Étape 2 : Le PBS s’hybride à sa séquence complémentaire sur le brin coupé. Étape 3 : La RT utilise le RTT comme modèle pour rétro transcrire le brin coupé. Étape 4 : Les mésappariements seront réparés par un « 5’ flap » ou un « 3’ flap ». (Figure adaptée de Godbout et al. [11]). pegRNA : Prime editing guide RNA ; PAM : protospacer adjacent motif ; PBS : primer binding site ; RTT : reverse transcriptase template. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.