")
")
| Issue |
Med Sci (Paris)
Volume 40, Number 5, Mai 2024
Nos jeunes pousses ont du talent !
|
|
|---|---|---|
| Page(s) | 467 - 470 | |
| Section | Partenariat médecine/sciences - Écoles doctorales - Masters | |
| DOI | https://doi.org/10.1051/medsci/2024047 | |
| Published online | 31 May 2024 | |
Une thérapie génique systémique dans le traitement de la mucoviscidose
A systemic gene therapy for the treatment of cystic fibrosis
Université de Tours, Master 2 infectiologie, immunité, vaccinologie et biomédicaments, Tours, France
* This email address is being protected from spambots. You need JavaScript enabled to view it.
** This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Le master I2VB se propose de donner les bases conceptuelles et pratiques des différents aspects de l’infectiologie. Il s’appuie sur une coopération exemplaire entre les équipes de recherche en infectiologie et en immunologie de l’Université de Tours, et celles, entre autres, de l’Unité Infectiologie et Santé Publique (ISP) du Centre INRAE de Tours-Nouzilly, concrétisée par une profonde interaction entre chercheurs et enseignants-chercheurs.
Cette formation aborde aussi bien les aspects fondamentaux et appliqués de l’infectiologie et de l’immunologie allant de l’étude moléculaire des interactions entre le pathogène et son hôte, jusqu’à la conception et la mise sur le marché des produits de la vaccinologie, des biothérapies anti-infectieuses et des anticorps immuno-thérapeutiques.
Le master I2VB (niveau M1) donne lieu aux parcours ICM, I&B et AcT (niveau M2).
-
L’option Infectiologie Cellulaire et Moléculaire (ICM) (responsables : Françoise Debierre-Grockiego et Martine Braibant) a pour objectifs de :
-
former des scientifiques dotés d’une culture générale et technique spécialisée dans les biotechnologies, l’infectiologie, les interactions hôte-pathogène et les mécanismes de la réponse immunitaire anti-infectieuse, contribuant à l’avancée des connaissances scientifiques et à ses applications industrielles, demandes sociétales en forte progression.
-
former des pharmaciens, médecins, vétérinaires, ingénieurs agronomes aux enjeux actuels de l’infectiologie à la fois dans les domaines fondamentaux et appliqués.
-
-
L’option Immunité et biomédicaments (I&B) (responsables : Anne di Tommaso et Isabelle Dimier-Poisson) a pour objectifs de :
-
former des scientifiques dotés d’une culture générale et technique spécialisée dans les biotechnologies, l’infectiologie, la vaccinologie, les biomédicaments et les biothérapies anti-infectieuses contribuant à l’avancée des connaissances scientifiques et à ses applications industrielles, demandes sociétales en forte progression.
-
former de jeunes scientifiques, pharmaciens, médecins, vétérinaires, ingénieurs agronomes aux enjeux actuels de l’infectiologie et des biomédicaments à la fois dans les domaines fondamentaux et appliqués.
-
-
L’option Anticorps thérapeutiques (AcT) (responsables : Laurie Lajoie et Isabelle Dimier-Poisson) a pour objectifs de :
-
former des scientifiques dotés d’une culture générale et technique spécialisée dans les biotechnologies, l’immunologie, la cancérologie et les biomédicaments dont les anticorps thérapeutiques, contribuant à l’avancée des connaissances scientifiques et à ses application industrielles et juridiques, demandes sociétales en forte progression.
-
former de jeunes scientifiques, pharmaciens, médecins, vétérinaires, ingénieurs agronomes aux enjeux actuels de l’infectiologie et des biomédicaments à la fois dans les domaines fondamentaux et appliqués.
-
© 2024 médecine/sciences – Inserm
L’actualité scientifique vue par les étudiants du Master Infectiologie, Immunologie, Vaccinologie & Biomédicaments (I2VB)
Équipe pédagogique
Françoise Debierre-Grockiego et Martine Braibant
Anne di Tommaso, Laurie Lajoie et Isabelle Dimier-Poisson

Série coordonnée par Sophie Sibéril.
La mucoviscidose est une maladie génétique rare à transmission autosomique et récessive, qui touche 1 naissance sur 3 500 en Europe [1]. Il est estimé que 2 millions de français sont porteurs sains du gène responsable de la maladie, c’est-à-dire possédant une seule copie du gène muté, contrairement aux sujets malades qui en portent deux. Le gène CFTR (cystic fibrosis transmembrane conductance regulator), situé sur le chromosome 7, code la protéine membranaire CFTR, un canal chlorure clé pour l’équilibre de la sécrétion et l’absorption d’ions et d’eau au niveau des tissus épithéliaux [2]. Chez les patients atteints de mucoviscidose, le gène CFTR est muté, ce qui entraîne un dysfonctionnement de la protéine [14] (→).
(→) Voir la Synthèse de E. Bardin et al., m/s n° 3, mars 2024, page 258
À ce jour, plus de 2 000 mutations du gène CFTR ont été recensées. Ces mutations sont classées selon leur nature et leurs conséquences fonctionnelles sur la protéine [3] (→).
(→) Voir la Synthèse de C. Férec, m/s n° 6-7, juin-juillet 2021, page 618
La plus courante, présente chez 70 % des malades, est la mutation F508del qui correspond à la délétion de 3 paires de bases au niveau du gène CFTR, et provoque des anomalies de repliement de la protéine et de son transport vers la membrane plasmique (mutation de classe II).
Le dysfonctionnement de la protéine CFTR provoque un épaississement du mucus sécrété par les cellules épithéliales, induit une augmentation du risque d’infections bactériennes et fongiques au niveau des poumons, ainsi qu’une diminution des fonctions pulmonaires, conduisant progressivement à une bronchopneumopathie chronique obstructive puis, à terme, à une insuffisance respiratoire [4]. D’autres organes sont touchés par ce dysfonctionnement, notamment le pancréas. Les enzymes produites par le pancréas restent bloquées dans le mucus épais de l’organe, ne pouvant pas rejoindre l’intestin, ce qui entraîne une altération du tissu pancréatique [5]. Les fonctions digestives des patients atteints de mucoviscidose sont également altérées, avec une malabsorption des graisses, des nutriments et des vitamines, associées à des douleurs abdominales, accompagnées de constipation ou de diarrhées.
Une prise en charge thérapeutique lourde pour les patients
En 2022, plus de 80 000 personnes étaient atteintes de mucoviscidose dans le monde. Depuis ces 20 dernières années, l’espérance de vie des patients s’est grandement améliorée, dépassant désormais l’âge de 50 ans [6]. Ceci a été possible grâce aux progrès de la recherche et à l’amélioration de la prise en charge des patients. Il est proposé aux patients des séances de kinésithérapie respiratoire, des entretiens avec des nutritionnistes, mais aussi la prise d’antibiotiques dans les cas d’infections pulmonaires. Cependant, face à la résistance aux antibiotiques, les impasses thérapeutiques sont de plus en plus fréquentes.
Le Kalydeco (ivacaftor) est un traitement de fond de la mucoviscidose prescrit chez les patients porteurs de l’une des mutations liées à un défaut de régulation du gène CFTR (classe III) [3]. Il s’agit d’un potentialisateur de la protéine CFTR, qui améliore son activité et ainsi la qualité du mucus sécrété. Mais ce traitement est inefficace chez les patients porteurs de la mutation F508del car il n’a aucun effet de correction sur le repliement et le transport de la protéine. En revanche, en association avec le Kalydeco, le Kaftrio (ivacaftor/tezacaftor/élexacaftor) peut être prescrit chez ces patients. Cette association augmente non seulement l’activité de la protéine CFTR, mais également sa quantité à la surface des cellules épithéliales.
Cependant, ces thérapies ne corrigent pas la mutation. Ce sont des traitements symptomatiques qui nécessitent une administration à vie, et dont les effets secondaires potentiels à long terme restent incertains. De plus, 10 % des patients ne peuvent pas bénéficier de cette prise en charge thérapeutique de par l’inefficacité sur certaines mutations [7].
La thérapie génique : la solution au problème ?
La thérapie génique est une stratégie thérapeutique indépendante de la nature des mutations. Elle convient donc à tous les patients atteints de la mucoviscidose. Elle permet une correction pérenne du gène ciblé et suscite donc beaucoup d’intérêt dans le cadre de la lutte contre cette maladie. À ce jour, l’utilisation de vecteurs viraux dérivés d’adénovirus a été testée mais n’a permis qu’une correction partielle de l’activité de la protéine CFTR et a engendré une inflammation exacerbée et destructrice [7].
Depuis 2012, l’édition de gènes par la méthode CRISPR/Cas9 est exploitée dans le cadre des maladies génétiques [8]. Le système CRISPR/Cas9 cible, grâce à un ARN guide, une séquence précise de l’ADN et coupe celle-ci via la fonction de ciseaux moléculaires de la Cas9 afin de remplacer un gène défectueux par un gène corrigé. Dans le cadre de la mucoviscidose, ce système a été testé pour corriger le gène CFTR défectueux [9]. Cependant, des effets hors cible provoquent des dommages dans le génome, rendant cette thérapie difficilement utilisable chez l’homme [10].
Une thérapie génique à grande échelle
Puisque la mucoviscidose est une maladie génétique systémique, il est nécessaire de développer un traitement qui va permettre d’effectuer une correction du gène défectueux dans toutes les cellules de l’organisme et en priorité dans celles affectées fonctionnellement. Bien que le système CRISPR/Cas9 soit un choix cohérent face à la problématique, sa distribution in vivo dans tout l’organisme reste difficile, la taille de la construction étant très importante. C’est dans ce contexte que Piotrowski-Daspit et al. ont récemment publié un article décrivant le développement d’une nanoparticule qui permet de remplacer la séquence d’ADN muté du gène CFTR pour corriger la mutation dans les cellules cibles [11]. Cette nanoparticule est composée de polymères de PLGA (poly lactic-co-glycolic acid) contenant une copie d’ADN simple brin donneur corrigée, ainsi qu’une séquence d’acides nucléiques peptidiques PNA (peptide nucleic acid) de structure tail-clamp (tcPNA) (Figure 1A). Cette molécule tcPNA est composée de bases puriques et pyrimidiques classiques et synthétiques, dont le squelette sucre-phosphate est remplacé par un squelette peptidique, ce qui confère une plus grande stabilité de liaison aux acides nucléiques endogènes que les bases classiques et ce, quelle que soit l’orientation de la séquence [12,13] (→).
(→) Voir le Dossier technique de F. Pellestor et al., m/s n° 6-7, juin-juillet 2005, page 753
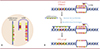 |
Figure 1. La nanoparticule PNA permet la correction de la mutation F508del du gène CFTR par des mécanismes de réparation de l’ADN. A. Structure de la nanoparticule PLGA (poly lactic-co-glycolic acid). La nanoparticule PLGA renferme l’ADN simple brin (ssADN) donneur portant le gène CFTR sain, ainsi que la tcPNA (tail clamp peptid nucleic acid). B. Mécanisme de correction du gène CFTR. La tcPNA forme une structure triplex PNA/ADN/PNA avec l’ADN cible à proximité du site de mutation du gène CFTR. Ceci déclenche le mécanisme de réparation de l’ADN médié par l’excision de nucléotides ce qui favorise la recombinaison homologue de l’ADN donneur au niveau du site muté du génome cible. Ainsi, la séquence mutée est remplacée par la séquence corrigée. |
Les chercheurs ont également ajouté des monomères de PNA sur lesquels a été greffé du polyéthylène glycol (PEG) en position γ (γPNA), permettant de renforcer la liaison à l’ADN.
Ainsi, cette molécule, créée pour avoir une complémentarité avec une séquence proche du gène CFTR, va pouvoir être absorbée dans les cellules par endocytose puis s’insérer au sein des deux brins d’ADN, formant une structure triple hélice (ou triplex) PNA/ADN/PNA (Figure 1B). L’ouverture des deux brins d’ADN déclenche alors un mécanisme de réparation endogène par excision de nucléotides et une recombinaison homologue avec la molécule d’ADN donneur, permettant ainsi son introduction dans le génome de la cellule.
C’est donc par ces mécanismes, que les chercheurs espèrent pouvoir corriger le gène CFTR muté dans toutes les cellules de l’organisme du patient atteint de mucoviscidose et ainsi permettre, à terme, de le guérir.
Des résultats prometteurs
Piotrowski-Daspit et al. ont dans un premier temps confirmé la diffusion des nanoparticules dans les organes prioritairement ciblés (poumons et intestins) suite à leur injection par voie intraveineuse à des souris homozygotes pour la mutation F508del.
Puis, les auteurs ont analysé la correction génotypique et phénotypique en développant un modèle ex vivo de cellules nasales épithéliales primaires, incubées en interface air-liquide (ALI). Après 3 ou 4 semaines de culture, les cellules ont été traitées par du PNA (trois fois, tous les deux jours). Ils ont ainsi pu observer par la technique de ddPCR (droplet digital PCR), technique fondée sur l’amplification d’une molécule d’ADN dans une gouttelette lipidique qui permet une quantification plus précise que les techniques de PCR conventionnelles, une augmentation significative de la quantité de séquences corrigées dans les cellules. Aussi, la mesure de la différence du courant de court-circuit (ΔIsc), par mise en place d’un courant électrique (permettant de démontrer la fonctionnalité des canaux CFTR) montre une augmentation significative du transport ionique dans ces mêmes cellules lorsqu’ils injectent les PNA, traduisant la bonne correction génotypique et phénotypique du gène CFTR.
Des résultats encore plus significatifs ont été obtenus avec un traitement par des γPNA, montrant une plus grande efficacité de correction. Étant plus prometteuse car offrant de meilleurs résultats, les auteurs ne se sont ensuite servis que de la nanoparticule γPNA.
Des tests ex vivo ont été effectués avec les cellules épithéliales nasales et intestinales isolées de souris modèle homozygotes, et ont démontré la correction phénotypique par l’étude du ΔIsc. De plus, une diminution significative de la quantité de cellules immunitaires dans le lavage broncho-alvéolaire des souris traitées par le γPNA par rapport au modèle non traité ou traité avec des γPNA vides, indique une baisse de l’inflammation dans les tissus.
Cette démonstration de la correction phénotypique dans le modèle ex vivo a également été observée in vivo, par l’analyse de la différence de potentiel nasal transépithélial, dans les cellules épithéliales nasales du modèle murin homozygote traitées avec les γPNA. En effet, les résultats indiquent une réponse des cellules nasales murines suite à une stimulation des canaux CFTR avec une solution sans chlore. La correction génotypique, elle, a été constatée de manière générale dans les cellules des voies respiratoires et intestinales des souris, et également du pancréas.
Enfin, cette thérapie n’a pas présenté d’effets indésirables, ni de toxicité détectable chez la souris. Aucun effet hors cible n’a été observé, traduisant la haute spécificité de correction de séquence permise grâce aux PNA.
Cette correction génotypique et phénotypique de la protéine CFTR de façon systémique est une avancée majeure dans la recherche de nouvelles thérapies contre la mucoviscidose. De par sa complexité de réalisation, aucune étude n’avait démontré cette possibilité jusqu’à ce jour, ce qui fait de cette nanoparticule, une potentielle thérapie innovante dans le cadre de cette maladie génétique. Afin d’optimiser cette méthode, des améliorations de la nanoparticule pourraient augmenter la durée des effets du traitement, ainsi que l’efficacité de transport du complexe. Une étude plus poussée concernant les paramètres de dosage et de fréquence d’administration permettraient de mieux comprendre et de contourner les limites de cette approche.
Un nouvel outil moléculaire pour les chercheurs
Le niveau des connaissances scientifiques ainsi que la facilité et le faible coût de la synthèse de l’ADN pourraient, dès aujourd’hui, permettre une production de ces nanoparticules à l’échelle industrielle. Elles permettraient d’améliorer la prise en charge des patients atteints de mucoviscidose mais pourraient également ouvrir la voie vers leur utilisation dans la recherche de traitement contre d’autres maladies génétiques monogéniques, voire multigéniques, affectant un type cellulaire spécifique dans un ou plusieurs organes. Dans un autre domaine, ces nanoparticules PNA pourraient également être utilisées en laboratoire de recherche et de développement comme outil de biologie moléculaire plus précis et plus sûr pour un ciblage des gènes à étudier, remplaçant le système CRISPR/Cas9 devenu aujourd’hui incontournable mais ayant comme inconvénient d’avoir des effets hors-cible.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Férec C, Scotet V. Genetics of cystic fibrosis: Basics. Archives de Pédiatrie 2020; 27 : eS4–7. [CrossRef] [Google Scholar]
- Ratjen F, Bell SC, Rowe SM, et al. Cystic fibrosis. Nat Rev Dis Primers 2015 ; 1 : 15010. [CrossRef] [Google Scholar]
- Férec C. La mucoviscidose: Du gène à la thérapeutique. Med Sci (Paris) 2021; 37 : 618–24. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Savant A, Lyman B, Bojanowski C, et al. Cystic Fibrosis. In: Adam MP, Everman DB, Mirzaa GM, et al., ed. GeneReviews®. Seattle (WA) : University of Washington, 2022. [Google Scholar]
- Ley D, Turck D. Digestive outcomes in Cystic fibrosis. Best Practice & Research Clinical Gastroenterology 2022; 56–7 : 101788. [CrossRef] [Google Scholar]
- Regard L, Martin C, Burnet E, et al. CFTR Modulators in People with Cystic Fibrosis : Real - World Evidence in France. Cells 2022; 11 : 1769. [CrossRef] [PubMed] [Google Scholar]
- Sui H, Xu X, Su Y, et al. Gene therapy for cystic fibrosis: Challenges and prospects. Front Pharmacol 2022; 13 : 1015926. [Google Scholar]
- Zhu Y. Advances in CRISPR/Cas9. Biomed Res Int 2022; 2022 : 9978571. [PubMed] [Google Scholar]
- Cabral B, Terlizzi V, Laselva O, et al. Anticipating New Treatments for Cystic Fibrosis: A Global Survey of Researchers. J Clin Med 2022; 11 : 1283. [CrossRef] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 2013 ; 31 : 822–826. [CrossRef] [PubMed] [Google Scholar]
- Piotrowski-Daspit AS, Barone C, Lin C-Y, et al. In vivo correction of cystic fibrosis mediated by PNA nanoparticles. Science Advances 2022; 8 : eabo0522. [CrossRef] [PubMed] [Google Scholar]
- Nielsen PE, Egholm M. An Introduction to Peptide Nucleic Acid. Current Issues in Molecular Biology 1999 ; 1 : 89–104. [PubMed] [Google Scholar]
- Pellestor F, Paulasova P, Macek M, et al. Les PNA (peptide nucleic acids): Des sondes high-tech pour l’analyse génétique et cytogénétique moléculaire. Med Sci (Paris) 2005 ; 21 : 753–758. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Bardin E, Pranke I, Hinzpeter A, Sermet-Gaudelus I. Traitements de la mucoviscidose. Révolution clinique et nouveaux défis. Med Sci (Paris) 2024; 40 : 258–67. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. La nanoparticule PNA permet la correction de la mutation F508del du gène CFTR par des mécanismes de réparation de l’ADN. A. Structure de la nanoparticule PLGA (poly lactic-co-glycolic acid). La nanoparticule PLGA renferme l’ADN simple brin (ssADN) donneur portant le gène CFTR sain, ainsi que la tcPNA (tail clamp peptid nucleic acid). B. Mécanisme de correction du gène CFTR. La tcPNA forme une structure triplex PNA/ADN/PNA avec l’ADN cible à proximité du site de mutation du gène CFTR. Ceci déclenche le mécanisme de réparation de l’ADN médié par l’excision de nucléotides ce qui favorise la recombinaison homologue de l’ADN donneur au niveau du site muté du génome cible. Ainsi, la séquence mutée est remplacée par la séquence corrigée. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.