")
")
| Issue |
Med Sci (Paris)
Volume 39, Number 10, Octobre 2023
|
|
|---|---|---|
| Page(s) | 754 - 762 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2023125 | |
| Published online | 09 November 2023 | |
Rôle du métabolisme cellulaire dans le contrôle des hépatites virales chroniques
Role of cellular metabolism in the control of chronic viral hepatitis
1
CIRI, Centre international de recherche en infectiologie, équipe VIRIMI, Univ Lyon, Inserm U1111, université Claude Bernard Lyon 1, CNRS, UMR5308, École normale supérieure (ENS) de Lyon, F-69007, Lyon, France
2
Service de virologie, hospices civils de Lyon, hôpital de la Croix-Rousse, Lyon, France
* Contribution égale This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les virus des hépatites modifient le métabolisme cellulaire des hépatocytes en interagissant avec des enzymes spécifiques, telles que la glucokinase. Les changements métaboliques induits par les virus peuvent avoir un impact direct sur la réponse antivirale innée. Les interactions complexes entre les composants viraux, l’immunité innée et le métabolisme des hépatocytes expliquent pourquoi les infections hépatiques chroniques conduisent à l’inflammation du foie, évoluant vers la cirrhose, la fibrose et le carcinome hépatocellulaire. Des régulateurs du métabolisme pourraient être utilisés dans des thérapies innovantes pour priver les virus de métabolites clés et induire une défense antivirale.
Abstract
Hepatitis viruses modify the cellular metabolism of hepatocytes by interacting with specific enzymes such as glucokinase. The metabolic changes induced by viruses can have a direct impact on the innate antiviral response. The complex interactions between viral components, innate immunity, and hepatocyte metabolism explain why chronic hepatitis infections lead to liver inflammation, progressing to cirrhosis, fibrosis, and hepatocellular carcinoma. Metabolic regulators could be used in innovative therapies to deprive viruses of key metabolites and induce an antiviral defense.
© 2023 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (© Laure Perrin-Cocon)
Les virus sont, en quelque sorte, des parasites intracellulaires qui dépendent du métabolisme de la cellule hôte pour leur réplication. Ils ont ainsi développé des mécanismes sophistiqués pour manipuler et moduler les voies de biosynthèse nécessaires à leurs besoins en énergie et en molécules élémentaires. Cela est particulièrement étudié pour les virus des hépatites qui établissent souvent des infections chroniques du foie, entraînant des troubles métaboliques et une inflammation chronique, à l’origine d’une fibrose du foie qui peut aboutir à la cirrhose, voire au carcinome hépatocellulaire (CHC).
Le foie est un organe central pour l’homéostasie métabolique et la réponse immunitaire. Les maladies métaboliques, comme l’obésité et la maladie du foie gras, se caractérisent par des défenses antivirales innées défectueuses et une inflammation chronique délétère. Des études récentes ont identifié des liens importants entre l’état métabolique des cellules et les fonctions de l’immunité innée, mais ces liens sont encore mal compris, notamment dans les hépatocytes. Cette interdépendance entre le métabolisme et l’immunité fait actuellement l’objet d’intenses recherches et suscite d’importants espoirs pour identifier des cibles permettant de contrôler la réplication virale.
Cet article de synthèse fait le point sur les connaissances des mécanismes d’interconnexion entre le métabolisme cellulaire et la défense innée antivirale. Il analyse comment les hépatites virales, causées par les virus des hépatites B, C et D (respectivement VHB, VHC et VHD), interfèrent avec le métabolisme des hépatocytes infectés afin de révéler des pistes pour le développement de nouvelles stratégies antivirales.
Ces trois virus hépatiques sont la cause d’hépatites chroniques mais ont des propriétés distinctes et présentent des différences majeures, notamment dans leur mode de réplication.
Le VHC est un virus à ARN monocaténaire de polarité positive appartenant à la famille des Flaviviridae. Il présente une grande diversité génétique avec plusieurs génotypes et sous-types différents. Lorsqu’il infecte l’hépatocyte, le VHC induit la formation d’un réseau membranaire intracellulaire connecté au réticulum endoplasmique, dont les circonvolutions hébergent les complexes de réplication virale. En interférant avec la voie de synthèse des lipoprotéines au niveau de ces structures, le VHC forme des particules fortement lipidées, les lipo-viro-particules (LVP), qui seront libérées par la voie de sécrétion vésiculaire (Figure 1A). Le VHC est une cause fréquente d’hépatite chronique et l’inflammation continue du foie qui en résulte entraîne une fibrose progressive. Celle-ci peut conduire à une cirrhose hépatique. Le carcinome hépatocellulaire est alors la complication la plus fréquente (13 % des cas) chez les patients présentant une hépatite C et une cirrhose [1].
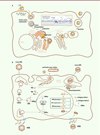 |
Figure 1. Schéma simplifié des cycles de réplication des VHC, VHB et VHD. A. Cycle de réplication du VHC. Les lipo-viro-particules (LVP) du VHC interagissent à la surface cellulaire avec différents cofacteurs permettant l’entrée du virus par endocytose. Après la fusion de l’enveloppe virale avec la membrane de l’endosome, la décapsidation libère le génome du virus dans la cellule. L’ARN viral est traduit en une polyprotéine qui est clivée en 10 protéines associées à la membrane du réticulum endoplasmique (RE) (voir encadré). La réplication du génome a lieu dans des sphérules membranaires dérivées du RE (réseau membranaire étroitement associé à des gouttelettes lipidiques [lipid droplets] cytoplasmiques). Lors de son bourgeonnement au travers de la membrane du RE, le virus emprunte la voie de biosynthèse des lipoprotéines de très faible densité (VLDL), se charge en apolipoprotéine (Apo) B et forme des particules riches en lipides neutres. Lors du processus de sécrétion, ces précurseurs se chargent en ApoE et ApoC pour être sécrétés sous la forme de LVP (lipo-viro particles). B. Cycle de réplication du VHB et de son virus satellite, le VHD. Suite à son interaction avec ses récepteurs cellulaires, en particulier le NTCP, la particule du VHB est internalisée dans les hépatocytes. La nucléocapside, contenant le génome viral sous forme circulaire relâché (ADNrc), est libérée dans la cellule et migre vers le noyau où l’ADNrc est réparé pour donner la forme circulaire close du génome, l’ADNccc, qui sert de matrice pour la transcription des différents ARNm codant les protéines virales. Parmi ces ARN, un ARN dit « pré-génomique » est encapsidé avec la polymérase virale. Cette dernière assure ensuite la synthèse d’un ADNrc grâce à son activité de transcriptase inverse. Au cours du bourgeonnement, la nucléocapside va acquérir les protéines d’enveloppe virale HBs avant d’être secrétée à l’extérieur de hépatocytes. Ces protéines HBs peuvent également être sécrétées sous forme de particules sous-virales, sphériques ou filamenteuses, non infectieuses. La particule du VHD porte à sa surface les enveloppes HBs du VHB, et utilise donc les mêmes récepteurs d’entrée dans la cellule. Comme pour le VHB, le génome viral du VHD est transporté au noyau où il recrute les ARN polymérases cellulaires. Celles-ci permettent d’une part la réplication du génome viral par un mécanisme de cercle roulant faisant intervenir un intermédiaire ARN dit « antigénomique », et, d’autre part, la synthèse d’un ARNm à partir d’un unique cadre de lecture (ORF, pour open reading frame) codant la forme courte S de l’antigène delta, nécessaire pour la réplication virale. Au cours de la réplication, certains ARN antigénomiques sont édités, permettant la synthèse à partir de la même ORF de la forme longue L de l’antigène delta qui est indispensable pour l’assemblage des virions. Les formes S et L de l’antigène delta vont former avec l’ARN génomique, les ribonucléoprotéines virales qui seront ensuite exportées dans le cytoplasme puis bourgeonneront en s’associant aux glycoprotéines d’enveloppe HBs du VHB. ADNccc : ADN circulaire clos de façon covalente ; ADNrc : ADN relâché circulaire ; ADNsb : ADN simple brin ; ARNm : ARN messager ; ARNpg : ARN prégénomique ; CLDN1 : claudine 1 ; EGFR : epidermal growth factor receptor ; HBc : protéine de capside (antigène C) de l’hépatite B ; HBs : antigène de surface (S) de l’hépatite B ; HS : heparan sulfate ; NTCP : sodium taurocholate cotransporting polypeptide ; OCLN : occludine ; Pol : polymérase virale ; SR-B1 : scavenger receptor B1 ; lipid droplets : gouttelettes lipidiques. |
Le VHB est un virus à ADN circulaire partiellement bicaténaire de la famille des Hepadnaviridae. Il infecte également les hépatocytes mais se réplique dans leur noyau (Figure 1B). Le VHB est connu pour sa capacité à établir une infection chronique. Il persiste en effet dans le noyau des hépatocytes sous la forme d’un mini-chromosome, le cccDNA (covalently closed circular DNA). Une autre particularité du VHB est sa capacité à produire un ARN précoce, appelé ARN pré-génomique, qui sert de matrice pour la synthèse de l’ADN viral (Figure 1B). L’infection chronique par le VHB est une cause majeure de carcinome hépatocellulaire. En plus d’induire une inflammation chronique et une nécrose associée à la réponse immunitaire, le VHB joue un rôle direct dans le développement du carcinome hépatocellulaire en induisant une instabilité génomique suite à l’intégration de son ADN dans le génome de l’hôte, et en affectant diverses fonctions cellulaires via des protéines virales [2].
Le VHD est un virus à ARN monocaténaire circulaire de polarité négative de la famille des Kolmioviridae et du genre Deltavirus. Contrairement aux autres virus hépatiques, le VHD ne peut pas se répliquer seul. Il nécessite la présence concomitante du VHB pour produire de nouveaux virions. En effet, lorsque le VHD infecte une cellule hépatique déjà infectée par le VHB, il utilise les protéines d’enveloppe du VHB pour produire de nouvelles particules virales. Le génome à ARN du VHD code deux isoformes d’une seule protéine, l’antigène delta, qui interagit avec le génome viral et les protéines d’enveloppe du VHB pour former des virions infectieux (Figure 1B). La surinfection par le VHD des patients chroniquement infectés par le VHB accroît le risque et accélère le développement de la cirrhose, favorisant le carcinome hépatocellulaire [3].
Les hépatites virales interfèrent avec le métabolisme de la cellule hôte
Si l’on sait depuis longtemps que le métabolisme cellulaire est modulé lors d’une infection virale, on commence seulement à comprendre à l’échelle moléculaire les mécanismes d’interférence entre infection virale et flux métabolique dans la cellule infectée. Toutes les cellules de l’organisme utilisent le glucose comme source de carbone et d’énergie, laquelle étant principalement échangée dans la cellule sous la forme d’adénosine triphosphate (ATP). Les hépatocytes, cellules spécialisées du foie, possèdent, en plus, la capacité de stocker ce glucose sous forme de glycogène lors des apports alimentaires, ou, au contraire, de le secréter pour maintenir la glycémie entre chaque repas. L’hépatocyte participe également à l’homéostasie lipidique, assurant la captation et le remodelage des lipides apportés par l’alimentation pour sécréter des lipoprotéines de faible densité (ou VLDL, pour very low density lipoprotein). Ces caractéristiques font du foie un organe central dans la régulation de l’homéostasie glucido-lipidique de l’organisme. L’ensemble des voies métaboliques impliquées dans ce processus est appelé métabolisme central du carbone (ou MCC) (Figure 2).
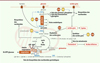 |
Figure 2. Impact des virus des hépatites sur le métabolisme central du carbone - liens avec la biosynthèse des nucléosides. Les cellules de l’organisme utilisent le glucose comme source de carbone et d’énergie. Il est d’abord phosphorylé par les hexokinases (HK ou GCK) et dégradé par la glycolyse en pyruvate. Le pyruvate peut alors être converti en lactate qui est excrété. Il peut également entrer dans la mitochondrie où il est transformé en acétyl-CoA, sous l’action de la PDH, et alimente ainsi le cycle de Krebs. La PDH est régulée par les kinases PDK. L’oxydation des acides gras peut également alimenter ce cycle en acétyl-CoA. Inversement, le citrate produit à partir d’acétyl-CoA peut sortir de la mitochondrie pour alimenter la synthèse des acides gras et des lipides. Une réaction anaplérotique, utilisant la glutamine et produisant un intermédiaire, permet d’équilibrer la synthèse d’alpha-keto-glutarate. Les enzymes (en bleu) ou voies métaboliques ciblées par les virus des hépatites ou leurs composants sont indiquées. ADP : adénosine diphosphate ; ATP : adénosine triphosphate ; CAD : carbamoyl-phosphate synthetase 2, aspartate transcarbamoylase, and dihydroorotase ; DHODH : dihydroorotate déshydrogénase ; FATP : transporteur d’acide gras ; GCK : glucokinase ; glucose-6-P : glucose-6-phosphate ; GLUT : transporteur de glucose ; HK : hexokinase ; MCT : transporteur de monocarboxylate ; PDH : pyruvate déshydrogénase ; PDK : pyruvate déshydrogénase kinase ; PKM2 : pyruvate kinase isoforme M2; SDH : succinate déshydrogénase. |
Les virus hépatiques ont développé des mécanismes de détournement du MCC au bénéfice de la réplication virale et de la production de nouveaux virions. Ainsi, le VHC et le VHB augmentent l’activité glycolytique de la cellule qu’ils infectent de différentes manières (Figure 2). Il a été en effet observé que le VHC provoque une chute de l’expression des protéines mitochondriales impliquées dans la phosphorylation oxydative [4]. En parallèle, la glycolyse est activée, ce qui permet de maintenir les niveaux intracellulaires d’ATP, mais aussi d’alimenter l’ensemble des voies anaboliques nécessaires à la réplication virale. Le VHC active également la glucokinase (GCK) et induit l’activité de la pyruvate déshydrogénase kinase (PDK), deux enzymes limitantes, à l’entrée et en aval de la glycolyse [5, 6]. Le virus est également capable d’induire l’expression d’enzymes de la gluconéogenèse, comme la phosphoénolpyruvate carboxykinase, ou de moduler la signalisation du récepteur de l’insuline, induisant ainsi une résistance de l’hépatocyte à cette hormone [7, 8].
Les glycoprotéines de surface S du VHB sont aussi impliquées dans l’activation de la glycolyse, en particulier leur forme longue qui affecte l’oligomérisation de l’isoforme M2 de la pyruvate kinase (PKM2), augmentant ainsi la glycolyse et la sécrétion de lactate [9] (Figure 2). Remarquablement, une simple transfection de la protéine de capside du VHB dans des cellules de carcinome hépatocellulaire induit une augmentation de la glycolyse, ce qui participerait au développement du cancer du foie [10].
Le VHC interfère également avec le métabolisme des lipides dans le foie. L’histoire naturelle de l’infection est en effet associée à une stéatose hépatique (accumulation de graisses dans le foie). Comme nous l’avons vu, le VHC induit d’abord la synthèse d’un réseau de membranes lipidiques intracellulaires constituant les usines de réplication virale [11]. Lors de sa morphogenèse, il interfère avec la voie de synthèse des VLDL pour former des particules hybrides contenant du matériel viral et lipoprotéique, les lipo-viro-particules [12, 13]. Ces particules sont une constante de l’infection chronique. Elles constituent une population hétérogène de structures formées de lipoprotéines modifiées, dont certaines ne contiennent que les enveloppes virales et un contenu très riche en lipides neutres [14], alors que d’autres, contenant en plus la capside virale, sont les particules circulantes les plus infectieuses [12]. Leur observation en microscopie électronique a permis de confirmer leur structure hybride constituée de lipoprotéines portant à leur surface les glycoprotéines d’enveloppe du virus [12, 15]. De nombreuses études ont montré que, lors de la réplication virale, différentes protéines du VHC peuvent induire la synthèse lipidique dans l’hépatocyte. On peut citer, par exemple, la protéine non structurale NS5A, dont l’expression augmente celle de la fatty acid synthase (FAS), une enzyme limitante de la synthèse des triglycérides [16] (Figure 2). Le VHB induit également des changements majeurs dans le métabolisme lipidique des hépatocytes. L’expression des gènes codant des protéines impliquées dans la synthèse des lipides est en effet augmentée chez des souris transgéniques qui répliquent le VHB dans le foie [17]. De même, la seule expression de la protéine X du VHB (HBx), une protéine régulatrice de la transcription virale, peut provoquer une stéatose hépatique ainsi qu’une augmentation de l’expression de la protéine FABP1 (fatty acid-binding protein 1), impliquée dans la liaison, le transport et le métabolisme des lipides [18] (Figure 2). Dans les mitochondries, l’oxydation des acides gras permet la production d’acétyl-CoA, alimentant le cycle de Krebs et donc la phosphorylation oxydative (OXPHOS) pour générer de grandes quantités d’ATP (Figure 2). Cette augmentation du métabolisme lipidique permettrait de maintenir la production d’ATP dans les cellules infectées soumises à un stress métabolique et favoriserait la carcinogenèse [19].
La défense antivirale innée et le métabolisme cellulaire sont interconnectés
Lors d’une infection, la mise en place de la réponse immunitaire innée repose sur la reconnaissance initiale de motifs spécifiques exprimés par les virus (ou PAMP, pour pathogen-associated molecular patterns) par des récepteurs cellulaires dédiés, les PRR (pathogen recognition receptors). Parmi ces PRR, les récepteurs de type Toll (ou TLR, pour Toll-like receptors) sont des récepteurs transmembranaires qui reconnaissent les PAMP présents dans le milieu extracellulaire ou qui ont été internalisés dans la cellule par endocytose. Les récepteurs de type RIG (RLR, pour RIG [retinoic acid inducible gene]-like receptors) ou STING (stimulator of interferon genes) sont quant à eux intracellulaires. Ils détectent les PAMP présents dans le cytoplasme de la cellule. La stimulation de ces PRR induit une signalisation intracellulaire conduisant à la production de cytokines inflammatoires et d’interférons (IFN) de type I et III1 contribuant à la défense antivirale (Figure 3).
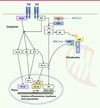 |
Figure 3. Voies de signalisation des récepteurs de l’immunité innée. Les récepteurs membranaires TLR humains activés recrutent les adaptateurs MyD88 (pour TLR1, 2, 4, 5, 6, 7, 8, 9) ou TRIF (pour TLR3 et 4), entraînant l’activation de NF-κB, de JNK, de p38-MAPK, et de TBK1/IKKε, conduisant, respectivement, à la sécrétion de cytokines pro-inflammatoires et d’interférons. L’activation de ces voies induit par ailleurs des changements métaboliques, notamment un switch glycolytique, nécessaires à la qualité de la réponse immunitaire innée ; dans les cellules dendritiques dérivées de monocytes humains, l’activation de p38-MAPK conduit à l’accumulation de HIF-1α, ce qui renforce l’expression d’enzymes de la glycolyse comme HK2. L’activation de AKT par TBK1 induit la phosphorylation de HK2, ce qui favorise sa liaison à la mitochondrie. Les ARN viraux sont détectés par les récepteurs cytosoliques de type RIG-I dont la signalisation repose sur la polymérisation de MAVS à la membrane mitochondriale. L’ADN viral est détecté par cGAS dont la signalisation dépend de STING. Ils déclenchent tous deux l’activation de TBK1/IKKε qui induit la sécrétion des interférons antiviraux. AKT : protéine kinase B (PKB) ; cGAS : cyclic GMP-AMP synthase ; HIF : hypoxia-induced factor ; IKKε : inhibitor of nuclear factor kappa B kinase subunit epsilon ; IRF : interferon response factor ; JNK : JUN N-terminal kinase ; MAPK : mitogen-activated protein kinase ; MAVS : mitochondrial antiviral-signaling protein ; MyD88 : MYD88 innate immune signal transduction adaptor ; NF-κB : nuclear factor kappa B ; RIG-I : retinoic acid inducible gene I ; STING : stimulator of interferon genes ; TBK1 : TANK-binding kinase 1 ; TLR : Toll-like receptor ; TRIF : TIR domain-containing adaptor molecule 1. |
La détection des PAMP par les PRR est à l’origine de profonds changements métaboliques qui sont nécessaires pour soutenir la mise en place de la réponse immunitaire innée. Ainsi, par exemple, l’activation de cellules dendritiques dérivées de monocytes par plusieurs TLR réoriente leur métabolisme vers la glycolyse [20–22]. Cette reprogrammation métabolique varie en fonction du type cellulaire considéré ; elle a cependant été peu étudiée dans les modèles hépatocytaires. Or, dans les hépatocytes primaires humains, le VHB est reconnu par le TLR2 [23] et des résultats obtenus dans les cellules de la lignée HepG2, une lignée cellulaire provenant du tissu hépatique d’un patient présentant un carcinome hépatocellulaire, suggèrent que l’engagement du TLR2 par le VHB pourrait, entre autres, induire une augmentation du métabolisme du cholestérol et une plus forte internalisation des VLDL [24]. De même, Chang et al. ont observé que les protéines Core et NS3 du VHC activent les macrophages humains via une signalisation impliquant le TLR2 associé au TLR1 ou au TLR6, et induisent la sécrétion de cytokines effectrices [25]. Les LVP de patients chroniquement infectés par le VHC altèrent également la réponse de cellules dendritiques à une stimulation par des ligands de TLR4 [26]. In vivo, chez la souris, l’infection par le virus de l’hépatite de la marmotte (WHV) provoque une augmentation de la glycolyse et une production accrue de lactate. Lors de cette infection par WHV, le TLR2 joue un rôle clé dans le remodelage métabolique des lymphocytes T CD8+ activés2, en favorisant la glycolyse et la glutaminolyse, ce qui serait essentiel à la mise en place de la réponse immunitaire cellulaire [27].
Les récepteurs de type RIG (RLR), tels que RIG-I (retinoic acid inducible gene I), sont des senseurs d’ARN viral cytosolique essentiels à la mise en place de la réponse antivirale innée [28, 29]. Or, la signalisation en aval de ces senseurs module le métabolisme cellulaire [30]. Réciproquement, l’hexokinase 2 (HK2), qui catalyse la première étape de la glycolyse, et le lactate, qui est le produit final de la glycolyse anaérobie, inhibent la signalisation par les RLR en interférant avec le recrutement par RIG-I de son adaptateur de signalisation MAVS (mitochondrial antiviral signaling protein) [31]. Ces mécanismes inhibiteurs seraient impliqués dans les hépatocytes infectés par le VHB, avec la formation d’un complexe associant HK2 à MAVS au niveau de la membrane mitochondriale, avec pour conséquence, l’inhibition de la signalisation en aval de RIG-I [32]. Outre ces interactions avec le métabolisme, de nombreuses études montrent que les virus VHB et VHC interfèrent directement avec les évènements de signalisation au niveau, ou en aval, des RLR. La protéine X et la polymérase du VHB, en se liant directement à MAVS, inhibent la réponse IFN, tandis que la protéine NS3-4A du VHC clive MAVS [33, 34]. Ces deux virus interfèrent aussi avec la détection d’ADN cytosolique par la voie STING, via cGAS (cyclic GMP-AMP synthase). Dans les hépatocytes primaires humains, la polymérase du VHB inhibe STING par interaction directe [35]. La protéine NS4B du VHC bloque également cette voie en inhibant l’interaction de STING avec son effecteur TBK1 (TANK-binding kinase 1) [36].
Les données disponibles, bien qu’encore très parcellaires, suggèrent ainsi que la reprogrammation métabolique induite par l’infection virale a un important impact sur la réponse immunitaire antivirale. Certaines protéines virales, en inhibant la signalisation en aval des PRR, et donc la réponse immunitaire innée, pourraient également altérer de façon indirecte l’état métabolique de la cellule.
Développement de nouvelles stratégies antivirales utilisant des régulateurs métaboliques
L’utilisation de médicaments ciblant le métabolisme des nucléosides pour limiter la réplication virale constitue une stratégie thérapeutique intéressante. En effet, la biosynthèse des nucléosides est connectée au métabolisme central du carbone, comme cela est montré Figure 2, notamment parce qu’elle utilise des intermédiaires produits par ces voies métaboliques. Leur modulation peut donc altérer la réplication virale qui en dépend. L’inhibition pharmacologique de l’inosine monophosphate déshydrogénase (IMPDH), de la voie de biosynthèse des purines, inhibe ainsi la réplication du VHC dans les cellules de la lignée hépatocytaire Huh7 et les modèles qui en sont dérivés [37]. De même, l’inhibition de la dihydroorotate déshydrogénase (DHODH), la quatrième enzyme de la voie de biosynthèse des pyrimidines (Figure 2), réprime la réplication du VHC dans les cellules Huh7.5 [38]. Ces effets antiviraux seraient liés à la fois à une chute du pool intracellulaire de nucléosides/nucléotides et à une induction de la réponse innée antivirale consécutive au stress cellulaire induit par ces traitements [39]. Il reste cependant à démontrer que ces observations, réalisées sur des cellules de lignées cancéreuses, sont transposables à des hépatocytes primaires ex vivo. En ce qui concerne la réplication du VHB, les inhibiteurs de biosynthèse des purines et des pyrimidines ont été testés, mais des effets opposés ont été décrits selon, notamment, le modèle étudié. Ainsi l’acide mycophénolique, un inhibiteur de l’IMPDH, augmente la réplication du VHB dans les cellules de lignées humaines cancéreuses d’origine hépatique, HepG2 et Huh7 [40,41], mais l’inhibe dans les hépatocytes primaires humains, qui constituent sans doute un modèle plus pertinent [42]. Chez des patients chroniquement infectés par le VHB et ayant reçu une greffe de foie, l’acide mycophénolique semble cependant n’avoir aucun effet sur la réplication virale, suggérant que les observations réalisées in vitro sur les hépatocytes primaires ne peuvent être transposées directement in vivo [43]. Enfin, il est à noter que l’inhibition de l’enzyme CAD (carbamoyl-phosphate synthetase 2, aspartate transcarbamoylase, and dihydroorotase), qui catalyse les trois premières étapes de la voie de biosynthèse des pyrimidines, en amont de la DHODH (Figure 2), bloque la réplication du VHD dans des hépatocytes primaires [44]. Néanmoins, à ce stade, il est encore trop tôt pour dire si ces observations, réalisées in vitro, sont transposables in vivo et peuvent déboucher sur des traitements efficaces pour les patients.
Une autre approche thérapeutique pour cibler l’infection par le VHB et le VHD pourrait reposer sur l’utilisation d’agonistes de FXR (farnesoid X receptor), un récepteur nucléaire formant des hétérodimères avec RXRa (retinoid X receptor alpha), exprimé notamment dans le foie et l’intestin, et jouant un rôle prépondérant dans la régulation de l’homéostasie des acides biliaires. En effet, ces deux virus dépendent indirectement du métabolisme des sels biliaires pour leur cycle réplicatif, car ils utilisent comme récepteur cellulaire la protéine NTCP (sodium taurocholate cotransporting polypeptide), un transporteur membranaire des acides biliaires fortement exprimé dans les hépatocytes humains [45]. L’expression de FXR est un facteur proviral du VHB et l’utilisation d’agonistes de ce récepteur inhibe la réplication du virus in vitro et in vivo [46]. Des modulateurs de FXR sont actuellement testés dans des essais cliniques comme inhibiteurs du VHB [47]. Certains agonistes de FXR inhibent la réplication du VHD mais aussi la sécrétion et l’infectiosité des particules virales [48]. En plus de jouer un rôle dans l’homéostasie des acides biliaires, FXR est étroitement impliqué dans le métabolisme des lipides et du cholestérol, régulant négativement leur synthèse et leur transport [49]. Les agonistes de FXR pourraient ainsi contrecarrer l’augmentation, induite par le VHB, de l’expression de gènes impliqués dans la synthèse des lipides, qui favorise le développement de la stéatose hépatique. Pour toutes ces raisons, l’utilisation de ligands de FXR semble être une approche prometteuse pour traiter l’infection par le VHB et le VHD ; en réduisant la réplication virale, elle pourrait réduire l’inflammation hépatique et prévenir les dommages associés à l’infection. Il a également été constaté qu’un agoniste spécifique du récepteur nucléaire RXRa inhibe l’infection par le VHB in vitro, alors que l’abaissement de son expression renforce l’infection virale. Cet effet inhibiteur sur l’infection précoce par le VHB semble dépendre d’une modulation des voies de synthèse de l’acide arachidonique et des eicosanoïdes [50].
Conclusion
En raison des liens étroits qui existent entre métabolisme, immunité innée et réplication virale dans le foie, l’étude des hépatites virales guide le développement de molécules antivirales agissant sur le métabolisme de l’hôte plutôt que directement sur les virus. Par rapport aux agents antiviraux à action directe, qui ciblent les composants viraux, les traitements qui touchent le métabolisme de l’hôte pour empêcher la réplication virale présentent plusieurs avantages, dont celui de limiter le risque d’émergence de virus résistants. Les modulations métaboliques induites par ces traitements peuvent, en outre, avoir un impact direct sur la réplication virale mais également stimuler la réponse innée antivirale via l’activation d’un ensemble de senseurs du stress métabolique.
Les voies de biosynthèse des lipides, du cholestérol et des sels biliaires sont des cibles thérapeutiques privilégiées dans le traitement des hépatites B et D, qui restent des problèmes de santé publique majeurs. Des résultats positifs lors d’essais cliniques permettraient d’établir une preuve de concept, ouvrant la voie à des thérapies antivirales innovantes fondées sur le ciblage du métabolisme cellulaire.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Financements
Ces travaux ont été financés par l’Agence nationale de recherches sur le Sida et les hépatites virales (ECTZ132153, ECTZ72972, ECTZ136480, ECTZ244976, ECTZ208004 et ECTZ208715), et l’Agence nationale de la recherche (22-CE15-0001).
Les interférons de type I comprennent les IFN-α et β, ubiquitaires, mais aussi les IFN-ω, ε et κ, dont l’action est limitée à certains organes. Le type II comprend uniquement l’IFN-γ, qui a principalement une fonction de communication entre cellules du système immunitaire. Le type III regroupe les IFN-λ qui induisent un état non spécialisé de résistance antivirale.
Les lymphocytes T CD8+ sont des cellules immunitaires jouant un rôle essentiel dans la lutte contre les virus, en éliminant les cellules qui les hébergent.
Références
- Nicoletti A, Ainora ME, Cintoni M, et al. Dynamics of liver stiffness predicts complications in patients with HCV related cirrhosis treated with direct-acting antivirals. Digestive and Liver Disease 2023; may 2 :S1590–8658(23)00579–0. [Google Scholar]
- Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol 2016 ; 64 : S84–S101. [CrossRef] [PubMed] [Google Scholar]
- Costante F, Stella L, Santopaolo F, et al. Molecular and Clinical Features of Hepatocellular Carcinoma in Patients with HBV-HDV Infection. J Hepatocell Carcinoma 2023; 10 : 713–24. [CrossRef] [Google Scholar]
- Diamond DL, Syder AJ, Jacobs JMet al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog 2010 ; 6 : e1000719. [CrossRef] [PubMed] [Google Scholar]
- Perrin-Cocon L, Kundlacz C, Jacquemin C, et al. Domain 2 of Hepatitis C Virus Protein NS5A Activates Glucokinase and Induces Lipogenesis in Hepatocytes. Int J Mol Sci 2022; 23 : 919. [CrossRef] [PubMed] [Google Scholar]
- Jung G-S, Jeon J-H, Choi Y-K, et al. Pyruvate dehydrogenase kinase regulates hepatitis C virus replication. Sci Rep 2016 ; 6 : 30846. [CrossRef] [PubMed] [Google Scholar]
- Shintani Y, Fujie H, Miyoshi H, et al. Hepatitis C virus infection and diabetes: direct involvement of the virus in the development of insulin resistance. Gastroenterology 2004 ; 126 : 840–848. [CrossRef] [PubMed] [Google Scholar]
- Hsieh M-J, Lan K-P, Liu H-Y, et al. Hepatitis C virus E2 protein involve in insulin resistance through an impairment of Akt/PKB and GSK3β signaling in hepatocytes. BMC Gastroenterol 2012 ; 12 : 74. [CrossRef] [PubMed] [Google Scholar]
- Wu Y-H, Yang Y, Chen C-H, et al. Aerobic glycolysis supports hepatitis B virus protein synthesis through interaction between viral surface antigen and pyruvate kinase isoform M2. PLoS Pathog 2021; 17 : e1008866. [CrossRef] [PubMed] [Google Scholar]
- Xie Q, Fan F, Wei W, et al. Multi-omics analyses reveal metabolic alterations regulated by hepatitis B virus core protein in hepatocellular carcinoma cells. Sci Rep 2017 ; 7 : 41089. [CrossRef] [PubMed] [Google Scholar]
- Blanchard E, Roingeard P. The Hepatitis C Virus-Induced Membranous Web in Liver Tissue. Cells 2018 ; 7 : 191. [CrossRef] [PubMed] [Google Scholar]
- Andre P, Komurian-Pradel F, Deforges S, et al. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J Virol 2002 ; 76 : 6919–6928. [CrossRef] [PubMed] [Google Scholar]
- Bartenschlager R, Penin F, Lohmann V, et al. Assembly of infectious hepatitis C virus particles. Trends Microbiol 2011 ; 19 : 95–103. [CrossRef] [PubMed] [Google Scholar]
- Scholtes C, Ramiere C, Rainteau D, et al. High plasma level of nucleocapsid-free envelope glycoprotein-positive lipoproteins in hepatitis C patients. Hepatology 2012 ; 56 : 39–48. [CrossRef] [PubMed] [Google Scholar]
- Piver E, Boyer A, Gaillard J, et al. Ultrastructural organisation of HCV from the bloodstream of infected patients revealed by electron microscopy after specific immunocapture. Gut 2017 ; 66 : 1487–1495. [CrossRef] [Google Scholar]
- Yang W, Hood BL, Chadwick SL, et al. Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology 2008 ; 48 : 1396–1403. [CrossRef] [PubMed] [Google Scholar]
- Hajjou M, Norel R, Carver R, et al. cDNA microarray analysis of HBV transgenic mouse liver identifies genes in lipid biosynthetic and growth control pathways affected by HBV. J Med Virol 2005 ; 77 : 57–65. [CrossRef] [PubMed] [Google Scholar]
- Wu Y-L, Peng X-E, Zhu Y-B, et al. Hepatitis B Virus X Protein Induces Hepatic Steatosis by Enhancing the Expression of Liver Fatty Acid Binding Protein. J Virol 2016 ; 90 : 1729–1740. [CrossRef] [PubMed] [Google Scholar]
- Wang M-D, Wu H, Huang S, et al. HBx regulates fatty acid oxidation to promote hepatocellular carcinoma survival during metabolic stress. Oncotarget 2016 ; 7 : 6711–6726. [CrossRef] [PubMed] [Google Scholar]
- Everts B, Amiel E, Huang SC, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol 2014 ; 15 : 323–332. [CrossRef] [PubMed] [Google Scholar]
- Perrin-Cocon L, Aublin-Gex A, Sestito SE, et al. TLR4 antagonist FP7 inhibits LPS-induced cytokine production and glycolytic reprogramming in dendritic cells, and protects mice from lethal influenza infection. Sci rep 2017 ; 7 : 40791. [CrossRef] [PubMed] [Google Scholar]
- Perrin-Cocon L, Aublin-Gex A, Diaz O, et al. Toll-like Receptor 4-Induced Glycolytic Burst in Human Monocyte-Derived Dendritic Cells Results from p38-Dependent Stabilization of HIF-1alpha and Increased Hexokinase II Expression. J Immunol 2018 ; 201 : 1510–1521. [CrossRef] [PubMed] [Google Scholar]
- Zhang Z, Trippler M, Real CI, et al. Hepatitis B Virus Particles Activate Toll-Like Receptor 2 Signaling Initially Upon Infection of Primary Human Hepatocytes. Hepatology 2020; 72 : 829–44. [CrossRef] [PubMed] [Google Scholar]
- Li Y-J, Zhu P, Liang Y, et al. Hepatitis B virus induces expression of cholesterol metabolism-related genes via TLR2 in HepG2 cells. World J Gastroenterol 2013 ; 19 : 2262–2269. [CrossRef] [PubMed] [Google Scholar]
- Chang S, Dolganiuc A, Szabo G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J Leukoc Biol 2007 ; 82 : 479–487. [CrossRef] [PubMed] [Google Scholar]
- Agaugue S, Perrin-Cocon L, andre P, et al. Hepatitis C lipo-Viro-particle from chronically infected patients interferes with TLR4 signaling in dendritic cell. PloS one 2007; 2 : e330. [Google Scholar]
- Zhang E, Ma Z, Li Q, et al. TLR2 Stimulation Increases Cellular Metabolism in CD8+ T Cells and Thereby Enhances CD8+ T Cell Activation, Function, and antiviral Activity. J Immunol 2019 ; 203 : 2872–2886. [CrossRef] [PubMed] [Google Scholar]
- Israelow B, Narbus CM, Sourisseau M, et al. HepG2 cells mount an effective antiviral interferon-lambda based innate immune response to hepatitis C virus infection. Hepatology 2014 ; 60 : 1170–1179. [CrossRef] [PubMed] [Google Scholar]
- Zhang Z, Filzmayer C, Ni Y, et al. Hepatitis D virus replication is sensed by MDA5 and induces IFN-β/λ responses in hepatocytes. J Hepatol 2018 ; 69 : 25–35. [CrossRef] [PubMed] [Google Scholar]
- Fekete T, Sütö MI, Bencze D, et al. Human Plasmacytoid and Monocyte-Derived Dendritic Cells Display Distinct Metabolic Profile Upon RIG-I Activation. Front Immunol 2018 ; 9 : 3070. [CrossRef] [PubMed] [Google Scholar]
- Zhang W, Wang G, Xu ZG, et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 2019 ; 178 : 176–89.e15. [CrossRef] [PubMed] [Google Scholar]
- Zhou L, He R, Fang P, et al. Hepatitis B virus rigs the cellular metabolome to avoid innate immune recognition. Nat Commun 2021; 12 : 98. [CrossRef] [PubMed] [Google Scholar]
- Wei C, Ni C, Song T, et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J Immunol 2010 ; 185 : 1158–1168. [CrossRef] [PubMed] [Google Scholar]
- Li K, Foy E, Ferreon JC, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A 2005 ; 102 : 2992–2997. [CrossRef] [PubMed] [Google Scholar]
- Liu Y, Li J, Chen J, et al. Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J Virol 2015 ; 89 : 2287–2300. [CrossRef] [PubMed] [Google Scholar]
- Ding Q, Cao X, Lu J, et al. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J Hepatol 2013 ; 59 : 52–58. [CrossRef] [PubMed] [Google Scholar]
- Pan Q, de Ruiter PE, Metselaar HJ, et al. Mycophenolic acid augments interferon-stimulated gene expression and inhibits hepatitis C Virus infection in vitro and in vivo. Hepatology 2012 ; 55 : 1673–1683. [CrossRef] [PubMed] [Google Scholar]
- Hoffmann H-H, Kunz A, Simon VA, et al. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc Natl Acad Sci U S A 2011 ; 108 : 5777–5782. [CrossRef] [PubMed] [Google Scholar]
- Wang Y, Wang W, Xu L, et al. Cross Talk between Nucleotide Synthesis Pathways with Cellular Immunity in Constraining Hepatitis E Virus Replication. antimicrob Agents Chemother 2016 ; 60 : 2834–2848. [CrossRef] [PubMed] [Google Scholar]
- Ruan J, Sun S, Cheng X, et al. Mitomycin, 5-fluorouracil, leflunomide, and mycophenolic acid directly promote hepatitis B virus replication and expression in vitro. Virol J 2020; 17 : 89. [CrossRef] [PubMed] [Google Scholar]
- Hoppe-Seyler K, Sauer P, Lohrey C, et al. The inhibitors of nucleotide biosynthesis leflunomide, FK778, and mycophenolic acid activate hepatitis B virus replication in vitro. Hepatology 2012 ; 56 : 9–16. [CrossRef] [PubMed] [Google Scholar]
- Gong ZJ, De Meyer S, Clarysse C, et al. Mycophenolic acid, an immunosuppressive agent, inhibits HBV replication in vitro. J Viral Hepat 1999 ; 6 : 229–236. [CrossRef] [PubMed] [Google Scholar]
- Ben-Ari Z, Zemel R, Tur-Kaspa R. The addition of mycophenolate mofetil for suppressing hepatitis B virus replication in liver recipients who developed lamivudine resistance–no beneficial effect. Transplantation 2001 ; 71 : 154–156. [CrossRef] [PubMed] [Google Scholar]
- Verrier ER, Weiss A, Bach C, et al. Combined small molecule and loss-of-function screen uncovers estrogen receptor alpha and CAD as host factors for HDV infection and antiviral targets. Gut 2020; 69 : 158–67. [CrossRef] [PubMed] [Google Scholar]
- Yan H, Zhong G, Xu G, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012 ; 1 : e00049. [CrossRef] [PubMed] [Google Scholar]
- Mouzannar K, Fusil F, Lacombe B, et al. Farnesoid X receptor-alpha is a proviral host factor for hepatitis B virus that is inhibited by ligands in vitro and in vivo. FASEB J 2019 ; 33 : 2472–2483. [CrossRef] [PubMed] [Google Scholar]
- Erken R, andre P, Roy E, et al. Farnesoid X receptor agonist for the treatment of chronic hepatitis B: A safety study. J Viral Hepat 2021; 28 : 1690–8. [CrossRef] [PubMed] [Google Scholar]
- Legrand A-F, Lucifora J, Lacombe B, et al. Farnesoid X receptor alpha ligands inhibit HDV in vitro replication and virion infectivity. Hepatol Commun 2023; 7 : e0078. [PubMed] [Google Scholar]
- Chiang JYL, Ferrell JM. Discovery of farnesoid X receptor and its role in bile acid metabolism. Mol Cell Endocrinol 2022; 548 : 111618. [CrossRef] [PubMed] [Google Scholar]
- Song M, Sun Y, Tian J, et al. Silencing Retinoid X Receptor Alpha Expression Enhances Early-Stage Hepatitis B Virus Infection In Cell Cultures. J Virol 2018 ; 92 : e01771–e01717. [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Schéma simplifié des cycles de réplication des VHC, VHB et VHD. A. Cycle de réplication du VHC. Les lipo-viro-particules (LVP) du VHC interagissent à la surface cellulaire avec différents cofacteurs permettant l’entrée du virus par endocytose. Après la fusion de l’enveloppe virale avec la membrane de l’endosome, la décapsidation libère le génome du virus dans la cellule. L’ARN viral est traduit en une polyprotéine qui est clivée en 10 protéines associées à la membrane du réticulum endoplasmique (RE) (voir encadré). La réplication du génome a lieu dans des sphérules membranaires dérivées du RE (réseau membranaire étroitement associé à des gouttelettes lipidiques [lipid droplets] cytoplasmiques). Lors de son bourgeonnement au travers de la membrane du RE, le virus emprunte la voie de biosynthèse des lipoprotéines de très faible densité (VLDL), se charge en apolipoprotéine (Apo) B et forme des particules riches en lipides neutres. Lors du processus de sécrétion, ces précurseurs se chargent en ApoE et ApoC pour être sécrétés sous la forme de LVP (lipo-viro particles). B. Cycle de réplication du VHB et de son virus satellite, le VHD. Suite à son interaction avec ses récepteurs cellulaires, en particulier le NTCP, la particule du VHB est internalisée dans les hépatocytes. La nucléocapside, contenant le génome viral sous forme circulaire relâché (ADNrc), est libérée dans la cellule et migre vers le noyau où l’ADNrc est réparé pour donner la forme circulaire close du génome, l’ADNccc, qui sert de matrice pour la transcription des différents ARNm codant les protéines virales. Parmi ces ARN, un ARN dit « pré-génomique » est encapsidé avec la polymérase virale. Cette dernière assure ensuite la synthèse d’un ADNrc grâce à son activité de transcriptase inverse. Au cours du bourgeonnement, la nucléocapside va acquérir les protéines d’enveloppe virale HBs avant d’être secrétée à l’extérieur de hépatocytes. Ces protéines HBs peuvent également être sécrétées sous forme de particules sous-virales, sphériques ou filamenteuses, non infectieuses. La particule du VHD porte à sa surface les enveloppes HBs du VHB, et utilise donc les mêmes récepteurs d’entrée dans la cellule. Comme pour le VHB, le génome viral du VHD est transporté au noyau où il recrute les ARN polymérases cellulaires. Celles-ci permettent d’une part la réplication du génome viral par un mécanisme de cercle roulant faisant intervenir un intermédiaire ARN dit « antigénomique », et, d’autre part, la synthèse d’un ARNm à partir d’un unique cadre de lecture (ORF, pour open reading frame) codant la forme courte S de l’antigène delta, nécessaire pour la réplication virale. Au cours de la réplication, certains ARN antigénomiques sont édités, permettant la synthèse à partir de la même ORF de la forme longue L de l’antigène delta qui est indispensable pour l’assemblage des virions. Les formes S et L de l’antigène delta vont former avec l’ARN génomique, les ribonucléoprotéines virales qui seront ensuite exportées dans le cytoplasme puis bourgeonneront en s’associant aux glycoprotéines d’enveloppe HBs du VHB. ADNccc : ADN circulaire clos de façon covalente ; ADNrc : ADN relâché circulaire ; ADNsb : ADN simple brin ; ARNm : ARN messager ; ARNpg : ARN prégénomique ; CLDN1 : claudine 1 ; EGFR : epidermal growth factor receptor ; HBc : protéine de capside (antigène C) de l’hépatite B ; HBs : antigène de surface (S) de l’hépatite B ; HS : heparan sulfate ; NTCP : sodium taurocholate cotransporting polypeptide ; OCLN : occludine ; Pol : polymérase virale ; SR-B1 : scavenger receptor B1 ; lipid droplets : gouttelettes lipidiques. |
| Dans le texte | |
|
Figure 2. Impact des virus des hépatites sur le métabolisme central du carbone - liens avec la biosynthèse des nucléosides. Les cellules de l’organisme utilisent le glucose comme source de carbone et d’énergie. Il est d’abord phosphorylé par les hexokinases (HK ou GCK) et dégradé par la glycolyse en pyruvate. Le pyruvate peut alors être converti en lactate qui est excrété. Il peut également entrer dans la mitochondrie où il est transformé en acétyl-CoA, sous l’action de la PDH, et alimente ainsi le cycle de Krebs. La PDH est régulée par les kinases PDK. L’oxydation des acides gras peut également alimenter ce cycle en acétyl-CoA. Inversement, le citrate produit à partir d’acétyl-CoA peut sortir de la mitochondrie pour alimenter la synthèse des acides gras et des lipides. Une réaction anaplérotique, utilisant la glutamine et produisant un intermédiaire, permet d’équilibrer la synthèse d’alpha-keto-glutarate. Les enzymes (en bleu) ou voies métaboliques ciblées par les virus des hépatites ou leurs composants sont indiquées. ADP : adénosine diphosphate ; ATP : adénosine triphosphate ; CAD : carbamoyl-phosphate synthetase 2, aspartate transcarbamoylase, and dihydroorotase ; DHODH : dihydroorotate déshydrogénase ; FATP : transporteur d’acide gras ; GCK : glucokinase ; glucose-6-P : glucose-6-phosphate ; GLUT : transporteur de glucose ; HK : hexokinase ; MCT : transporteur de monocarboxylate ; PDH : pyruvate déshydrogénase ; PDK : pyruvate déshydrogénase kinase ; PKM2 : pyruvate kinase isoforme M2; SDH : succinate déshydrogénase. |
| Dans le texte | |
|
Figure 3. Voies de signalisation des récepteurs de l’immunité innée. Les récepteurs membranaires TLR humains activés recrutent les adaptateurs MyD88 (pour TLR1, 2, 4, 5, 6, 7, 8, 9) ou TRIF (pour TLR3 et 4), entraînant l’activation de NF-κB, de JNK, de p38-MAPK, et de TBK1/IKKε, conduisant, respectivement, à la sécrétion de cytokines pro-inflammatoires et d’interférons. L’activation de ces voies induit par ailleurs des changements métaboliques, notamment un switch glycolytique, nécessaires à la qualité de la réponse immunitaire innée ; dans les cellules dendritiques dérivées de monocytes humains, l’activation de p38-MAPK conduit à l’accumulation de HIF-1α, ce qui renforce l’expression d’enzymes de la glycolyse comme HK2. L’activation de AKT par TBK1 induit la phosphorylation de HK2, ce qui favorise sa liaison à la mitochondrie. Les ARN viraux sont détectés par les récepteurs cytosoliques de type RIG-I dont la signalisation repose sur la polymérisation de MAVS à la membrane mitochondriale. L’ADN viral est détecté par cGAS dont la signalisation dépend de STING. Ils déclenchent tous deux l’activation de TBK1/IKKε qui induit la sécrétion des interférons antiviraux. AKT : protéine kinase B (PKB) ; cGAS : cyclic GMP-AMP synthase ; HIF : hypoxia-induced factor ; IKKε : inhibitor of nuclear factor kappa B kinase subunit epsilon ; IRF : interferon response factor ; JNK : JUN N-terminal kinase ; MAPK : mitogen-activated protein kinase ; MAVS : mitochondrial antiviral-signaling protein ; MyD88 : MYD88 innate immune signal transduction adaptor ; NF-κB : nuclear factor kappa B ; RIG-I : retinoic acid inducible gene I ; STING : stimulator of interferon genes ; TBK1 : TANK-binding kinase 1 ; TLR : Toll-like receptor ; TRIF : TIR domain-containing adaptor molecule 1. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.