")
")
| Issue |
Med Sci (Paris)
Volume 38, Decembre 2022
Les Cahiers de Myologie
|
|
|---|---|---|
| Page(s) | 46 - 48 | |
| Section | Cas clinique | |
| DOI | https://doi.org/10.1051/medsci/2022178 | |
| Published online | 16 January 2023 | |
La grande variabilité phénotypique des mutations du gène RYR1
The high phenotypic variability of RYR1 gene mutations
1
Service neurologie, CHU Mustapha Pacha Alger, Algérie
2
Service neurologie, EHS Cherchell Tipaza, Algérie
3
Unité de myogénétique, Hôpital Pitié Salpêtrière Paris, France
4
Département biochimie et génétique moléculaire, CHU Grenoble, France
* This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Le gène RYR1 (Ryanodine-Receptor-1) code pour une protéine-clé dans le processus de couplage excitation-contraction de la fibre musculaire. Ce récepteur est le principal canal de libération du calcium à partir du réticulum endoplasmique [1]. Un certain nombre de phénotypes cliniques sont imputables aux mutations de ce gène de grande taille comme rappelé dans la liste établie par ORPHANET (voir Encadré). Nous décrivons, dans ce travail, deux phénotypes distincts, et trompeurs à certains égards, en rapport avec des mutations de ce gène.
Abstract
The RYR1 gene encodes the ryanodine-receptor 1, a key protein in the excitation-contraction coupling that takes place in muscle fibers. This receptor is the main channel responsible for calcium release from the endoplasmic reticulum [1]. A number of clinical phenotypes are linked to various mutations in this large gene as shown in a compilation established by ORPHANET (see table). In this work we describe two distinct, somewhat misleading, phenotypes in relation to pathogenic variants in this gene.
© 2022 médecine/sciences – Inserm

© Bertrand Jordan
Observation n° 1
Dans la première famille (famille 1, Figure 1), le propositus présentait un phénotype ayant fait évoquer une forme pseudomyopathique de syndrome myasthénique congénital (SMC). Le patient était le seul atteint au sein d’une fratrie de huit enfants issus d’un couple algérien consanguin. Comme indiqué sur l’arbre généalogique, d’autres membres de la parentèle pourraient être atteints mais ils n’ont pas été examinés. L’histoire clinique du patient index était marquée par une hypotonie néonatale suivie d’une ptose palpébrale fluctuante sur plusieurs semaines, ainsi qu’une fatigabilité musculaire très lentement progressive. Cette fatigabilité apparue vers de l’âge de 7 ans pouvait s’aggraver de manière nettement fluctuante dans la journée, et parfois même sur plusieurs semaines.
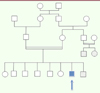 |
Figure 1. Famille 1 (phénotype SMC). Les sujets représentés en grisé auraient un phénotype superposable mais n’ont pas été examinés. |
Sur le plan fonctionnel, le périmètre de marche était réduit à 300 mètres et le patient présentait des difficultés à se relever du sol (signe de Gowers positif). L’examen clinique révélait une ophtalmoplégie complète avec ptosis bilatéral, un déficit des muscles du visage y compris au niveau des orbiculaires des yeux, une dysphagie aux liquides, et enfin un déficit moteur aux quatre membres prédominant en proximal et à l’origine d’une démarche dandinante. Le score myasthénique était à 55/100. Les données paracliniques sont résumées dans le tableau comparatif (Tableau I). L’ENMG était myopathique avec présence d’un décrément franc. Une atrophie des fibres de type II a été retrouvée sur la biopsie musculaire, comme c’est souvent le cas dans les SMC [2]. Sur les bases de la clinique et des données de laboratoire, le diagnostic de SMC a été retenu et a conduit à la mise en route d’un traitement symptomatique à base de pyridostigmine. Certains signes cliniques se sont améliorés : le périmètre de marche est passé à 500 mètres et le score myasthénique a atteint 65/100. L’étude génétique a permis d’identifier un variant pathologique à l’état homozygote situé dans l’exon 79 du gène RYR1. Cette mutation p.R3772W a déjà été rapportée dans la littérature [3] en lien avec un phénotype de myopathie congénitale (MC) et une susceptibilité à l’hyperthermie maligne (HM).
Comparatif des deux phénotypes (NA: non analysé).
Observation n° 2
Les patientes de la famille 2 sont deux sœurs d’origine palestinienne issues d’une union consanguine (famille 2, Figure 2). Leur phénotype était en faveur d’une myopathie congénitale avec un déficit moteur proximal, précoce et non fluctuant, présent aux quatre membres. Une scoliose sévère avec retentissement respiratoire était également notée. Leurs données paracliniques sont résumées dans le tableau comparatif. On retiendra le caractère myopathique de l’EMG, la présence de cores sur la biopsie musculaire et une augmentation significative des enzymes musculaires (à six fois la normale). Une mutation homozygote de l’exon 40 du gène RYR1 a été retrouvée dans cette famille. Ce variant référencé p.T2206M est associé à une susceptibilité à l’HM [4]. Une liste des différents anesthésiques contre-indiqués a été remise aux deux patientes afin de prévenir ce risque majeur potentiellement létal.
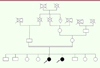 |
Figure 2. Famille 2 (phénotype de myopathie congénitale). |
Commentaire
Ces deux familles porteuses de variants pathologiques du gène RYR1 présentent des phénotypes sensiblement différents. La famille 1 a un phénotype très évocateur de SMC associé à des signes myopathiques tandis que la famille 2 se présente avec un tableau myopathique net et franc, sans fluctuation. Le tableau comparatif illustre bien ces différences phénotypiques.
La même mutation RYR1 que celle identifiée dans la famille 1 a déjà été rapportée chez trois patients issus de deux familles américaines non-apparentées [3]. Leur phénotype est parfaitement superposable à celui de notre observation. La biopsie avait également mis en évidence une atrophie des fibres de type II, sans présence de cores. Seule différence notable : l’absence de fluctuation des symptômes et la normalité de l’ENG. Dans cette famille, aucun traitement par pyridostigmine n’a été tenté. L’éventail phénotypique des mutations RYR1 pourrait donc, avec ces observations, s’élargir en direction des SMC. On note également que cette même mutation peut se transmettre selon un mode autosomique dominant et n’entraîne qu’une HM isolée [5]. Des phénotypes sans atteinte myopathique majeure ont par ailleurs été décrits en lien avec d’autres mutations de ce gène [6, 7].
Dans la série d’AlBakry [6], deux patients avaient la même mutation que celle de la famille 2 sans atteinte myopathique nette mais avec une ophtalmoplégie. Ce dernier signe n’avait pas été retrouvé dans notre famille (famille 2), ce qui illustre la variabilité phénotypique de cette mutation capable de donner aussi bien un phénotype SMC qu’un phénotype de myopathie congénitale. Ces éléments étendent le spectre phénotypique des mutations du gène aussi grand que le gène RYR1 est très étendu ; la susceptibilité accrue à l’HM reste toutefois leur point commun [8]. La notion de « ryanodinopathies » tend à s’imposer et permet d’englober les myopathies congénitales de type central core disease, la myopathie à minicore et certaines affections congénitales neuromusculaires [9]. La réponse positive de notre patient à la pyridostigmine est également retrouvée chez des patients présentant des phénotypes SMC mais avec une histologie de MC [10]. Deux autres patients présentant un phénotype SMC typique et similaire au nôtre ont été rapportés [11]. Leur réponse thérapeutique à la pyridostigmine était là aussi frappante et une mutation non-sens du gène RYR1 a été incriminée (p.Arg2241*). Il s’agissait d’un phénotype myasthenic-like dont la génétique a permis de poser un diagnostic de certitude de MC.
Des anomalies de la jonction neuromusculaire (JNM) peuvent être présentes dans les myopathies congénitales. Chez certains patients atteints de MC, des régions post-synaptiques rudimentaires et un déficit en récepteurs à l’acétylcholine ont été retrouvés. La neurotransmission peut s’en trouver compromise et la réponse post-synaptique amoindrie [12]. De plus, d’autres gènes responsables de MC peuvent aussi entraîner des anomalies fonctionnelles de la JNM et des récepteurs qui s’y trouvent. Elles sont plutôt incriminées dans des formes plutôt létales d’arthrogrypose congénitale et d’akinésie fœtale [13]. Par ailleurs, un séquençage complet du gène RYR1 entrepris chez 39 familles avec épisodes de rhabdomyolyse (avec ou sans myalgies), a démontré que 14 d’entre elles portaient des mutations de ce gène. La plupart des personnes concernées ne présentaient pas de faiblesse musculaire et n’avaient pas d’antécédents d’HM [14]. Ceci illustre parfaitement l’expansion des phénotypes associés à ce gène de très grande taille.
Les variants du gène RYR1 (même si la protéine codée ne fait pas partie stricto sensu des protéines de la JNM) doivent être recherchés devant tout phénotype myasthenic-like avec ou sans réponse positive au traitement par la pyridostigmine [15], et ce d’autant plus que les gènes des SMC auront été préalablement éliminés.
Phénotypes associés au gène RYR1 – Source Orphanet 2020
Mutation germinale causale dans Forme létale du syndrome des ptérygiums multiples ORPHA:33108
Mutation germinale causale dans Hyperthermie maligne de l’anesthésie ORPHA:423
Mutation germinale causale dans Myopathie à multi-minicores avec ophtalmoplégie externe ORPHA:98905
Mutation germinale causale dans Myopathie à multi-minicores modérée avec atteinte des mains ORPHA:178145
Mutation germinale causale dans Myopathie centronucléaire autosomique récessive ORPHA:169186
Mutation germinale causale dans Myopathie congénitale à « central cores » ORPHA:597
Mutation germinale causale dans Myopathie congénitale à début pseudo-myasthénique ORPHA:424107
Mutation germinale causale dans Myopathie congénitale bénigne des Samaritains ORPHA:324581
Mutation germinale causale dans Syndrome de King-Denborough ORPHA:99741
Facteur de susceptibilité majeur dans Hyperthermie maligne induite par l’exercice ORPHA:466650
Gène candidat testé dans Myopathie centronucléaire autosomique dominante ORPHA:169189
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Laver DR. Regulation of the RYR channel gating by Ca2+ and Mg2. Biophys Rev 2018 ; 10 : 1087–1095. [CrossRef] [PubMed] [Google Scholar]
- Maggi L, Bernasconi P, D’Amico A, et al. Italian recommendations for diagnosis and management of congenital myasthenic syndromes. Neurol Sci 2019 ; 40 : 457–468. [CrossRef] [PubMed] [Google Scholar]
- Shaaban S, Ramos-Platt L, Gilles FH, et al. RYR1 mutations as a cause of ophthalmoplegia, facial weakness, and malignant hyperthermia. JAMA Ophthalmol 2013 ; 131(12):1532–1540. [CrossRef] [PubMed] [Google Scholar]
- Monnier N, Krivosic-Horber R, Payen JF, et al. Presence of two different genetic traits in malignant hyperthermia families: implication for genetic analysis, diagnosis, and incidence of malignant hyperthermia susceptibility. Anesthesiology 2002 ; 97 : 1067–1074. [CrossRef] [PubMed] [Google Scholar]
- Levano S, Vukcevic M, Singer M, et al. Increasing the number of diagnostic mutations in malignant hyperthermia. Hum Mutat 2009 ; 30 : 590–598. [CrossRef] [PubMed] [Google Scholar]
- AlBakri A, Karaoui M, Alkuraya FS, et al. Congenital ptosis, scoliosis, and malignant hyperthermia susceptibility in siblings with recessive RYR1 mutations. J AAPOS 2015 ; 19 : 577–579. [CrossRef] [PubMed] [Google Scholar]
- Dilaver N, Mazaheri N, Maroofian R, et al. Novel homozygous missense mutation in RYR1 leads to severe congenital ptosis, ophthalmoplegia, and scoliosis in the absence of myopathy. Mol Syndromol 2017 ; 9 : 25–29. [Google Scholar]
- Rosenberg H, Pollock N, Schiemann A, et al. Malignant hyperthermia: a review. Orphanet J Rare Dis 2015 ; 10 : 93. [CrossRef] [PubMed] [Google Scholar]
- Alkhunaizi E, Shuster S, Shannon P, et al. Homozygous/compound heterozygote RYR1 gene variants: Expanding the clinical spectrum. Am J Med Genet A 2019 ; 179 : 386–396. [CrossRef] [PubMed] [Google Scholar]
- Robb SA, Sewry CA, Dowling JJ, et al. Impaired neuromuscular transmission and response to acetylcholinesterase inhibitors in centronuclear myopathies. Neuromuscul Disord 2011 ; 21 : 379–386. [CrossRef] [PubMed] [Google Scholar]
- Illingworth MA, Main M, Pitt M, et al. RYR1-related congenital myopathy with fatigable weakness, responding to pyridostigimine. Neuromuscul Disord 2014 ; 24 : 707–712. [CrossRef] [PubMed] [Google Scholar]
- Liewluck T, Shen XM, Milone M, et al. Endplate structure and parameters of neuromuscular transmission in sporadic centronuclear myopathy associated with myasthenia. Neuromuscul Disord 2011 ; 21 : 387–395. [CrossRef] [PubMed] [Google Scholar]
- Beecroft SJ, Lombard M, Mowat D, et al. Genetics of neuromuscular fetal akinesia in the genomics era. J Med Genet 2018 ; 55 : 505–514. [CrossRef] [PubMed] [Google Scholar]
- Colombo I, Scoto M, Manzur AY, et al. Congenital myopathies: Natural history of a large pediatric cohort. Neurology 2015 ; 84 : 28–35. [CrossRef] [PubMed] [Google Scholar]
- Lawal TA, Todd JJ, Meilleur KG. Ryanodine Receptor 1-related myopathies: diagnostic and therapeutic approaches. Neurotherapeutics 2018 ; 15 : 885–899. [CrossRef] [PubMed] [Google Scholar]
Liste des tableaux
Liste des figures
|
Figure 1. Famille 1 (phénotype SMC). Les sujets représentés en grisé auraient un phénotype superposable mais n’ont pas été examinés. |
| Dans le texte | |
|
Figure 2. Famille 2 (phénotype de myopathie congénitale). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.