")
")
| Issue |
Med Sci (Paris)
Volume 36, Number 6-7, Juin–Juillet 2020
Rétine
|
|
|---|---|---|
| Page(s) | 607 - 615 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2020095 | |
| Published online | 02 July 2020 | |
La thérapie génique des rétinites pigmentaires héréditaires
Gene therapy for retinitis pigmentosa
1
Centre hospitalier universitaire de Nantes, Nantes Université, service d’ophtalmologie, 1 place Alexis Ricordeau, 44093 Nantes, France
2
Inserm UMR 1089, thérapie génique translationnelle des maladies génétiques, IRS 2 - Nantes Biotech, 22 boulevard Benoni Goullin, 44200 Nantes, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les rétinites pigmentaires, ou dystrophies rétiniennes héréditaires, sont des maladies dégénératives cécitantes d’origine génétique. La thérapie génique est une approche révolutionnaire en plein essor qui ouvre la voie au traitement de maladies jusqu’ici incurables. Une thérapie génique, le Luxturna®, a obtenu une autorisation de mise sur le marché par la FDA (Food and Drug Administration) fin 2017 et l’EMA (European Medicines Agency) fin 2018. Ce traitement, à l’efficacité démontrée, destiné aux patients porteurs d’une amaurose congénitale de Leber ou d’une rétinopathie pigmentaire en lien avec une mutation bi-allélique du gène RPE65, apporte beaucoup plus de questions que de réponses. Nous présentons, dans cette revue, les avancées actuelles, puis les défis technologiques, économiques et éthiques à surmonter pour que la thérapie génique améliore nos pratiques médicales.
Abstract
Retinitis pigmentosa is the most common blinding inherited retinal dystrophy. Gene therapy is a burgeoning revolutionary approach that paves the way to treatment of previously incurable diseases. At the end of 2017 and 2018, a gene therapy, Luxturna®, obtained a marketing authorization by respectively the FDA (Food and Drug Administration) and the EMA (European Medicines Agency). This treatment, with proven efficacy, is available to patients with Leber congenital amaurosis and retinitis pigmentosa associated with bi-allelic mutations of the RPE 65 gene. In this paper, we present the current advances in gene therapy for retinitis pigmentosa and discuss the technological, economic and ethical challenges to overcome for gene therapy to improve medical practices.
© 2020 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (Photo © Inserm - Marion Vincent).
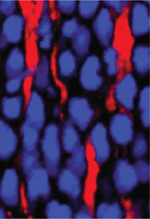
La rétine est le tissu neurosensoriel tapissant le fond de l’œil. La transformation du signal lumineux en un signal électrique est un processus complexe mettant en jeux de nombreux types cellulaires rétiniens (Figure 1). Le photorécepteur, activé par l’influx lumineux, transmet l’information visuelle aux cellules bipolaires qui intègrent le signal, avant de le transmettre aux cellules ganglionnaires, dont les axones constituent le nerf optique. Deux types de photorécepteurs co-existent : les bâtonnets, disséminées sur tout le fond de l’œil, assurent la vision périphérique. Leur seuil d’activation permet la vision en basse luminosité ; les cônes, concentrés dans la macula, assurent l’acuité visuelle, la stéréoscopie et la vision des couleurs. Situé entre les photorécepteurs et la choroïde, l’épithélium pigmentaire (EP) est une couche monocellulaire en contact étroit avec les segments externes des photorécepteurs via leurs microvillosités. L’EP joue un rôle de barrière hémato-rétinienne, de nutrition et de régénération des photorécepteurs, et dans le renouvellement du 11-cis retinal1 (étape indispensable du cycle visuel).
 |
Figure 1. Structure de la rétine en microscopie confocale. RPE : épithélium pigmentaire ; POS : segment externe des photorécepteurs ; PR : corps cellulaires des photorécepteurs ; HC : cellules horizontales ; BP : cellules bipolaires ; AC : cellules amacrines ; GC : cellules ganglionnaires. |
Les rétinites pigmentaires
Les rétinites pigmentaires (RP) sont des maladies rétiniennes dégénératives héréditaires qui constituent un ensemble génétiquement hétérogène impliquant plus de 90 gènes2. Elles affectent 1 personne sur 4 000 dans le monde. Elles sont sporadiques dans 30 % des cas et se transmettent le plus souvent de façon autosomique récessive (dans 50-60 % des cas), parfois autosomique dominante (30 à 40 % des cas) ou rarement liée à l’X (5 à 15 % des cas) [1]. Les mutations altèrent le plus souvent les photorécepteurs, parfois les cellules de l’EP. Elles mènent toutes, à plus ou moins long terme, à la destruction des deux types cellulaires et à la perte de la fonction visuelle. La baisse de vision se manifeste en général à l’âge adulte par une héméralopie3 puis une baisse du champ visuel périphérique (due à la perte des bâtonnets), et tardivement, par une baisse de l’acuité visuelle et de la vision des couleurs (due à la perte des cônes) : c’est la forme habituelle de RP de l’adulte ou « rod-cone » (pour bâtonnet et cône). L’atteinte dans l’ordre inverse, « cone-rod », est plus rare. Si l’atteinte est très précoce, elle altère le développement visuel du nourrisson et se manifeste par une malvoyance et un nystagmus4 dans les six premiers mois de vie : on parle alors d’amaurose5 congénitale de Leber (ACL). Il existe cependant un continuum entre ces deux phénotypes, car des enfants peuvent présenter une forme intermédiaire entre l’ACL et la RP de l’adulte : c’est la early onset severe retinal dystrophy (EOSRD). L’atteinte est uniquement oculaire dans 80 % des cas, mais elle peut aussi s’intégrer dans un ensemble syndromique (ciliopathies, dont les syndromes d’Usher, ou certaines maladies métaboliques). Ces mladies sont cécitantes et aucun traitement pharmacologique n’est actuellement disponible pour les guérir ou même stopper leur évolution.
Les principes de la thérapie génique
La thérapie génique est une stratégie innovante particulièrement intéressante dans les RP. Elle vise à restaurer la fonction moléculaire déficiente en transférant dans les cellules cibles un gène thérapeutique. L’apport du gène peut suivre plusieurs stratégies distinctes : supplémentation, correction, inactivation, inactivation et supplémentation, compensation. La supplémentation (apport du gène défaillant) est particulièrement adaptée pour les maladies à transmission récessive. La correction et l’inactivation d’un gène pathogène est plus pertinente en cas de maladie à transmission dominante. La compensation correspond, quant à elle, à l’apport d’un gène différent de celui qui est déficient afin de compenser la perte fonctionnelle. Cette approche est particulièrement adaptée si le gène malade n’est pas connu ou si la maladie est à un stade avancé. En effet, le transfert de gènes thérapeutiques n’a de sens que si les cellules ciblées sont encore présentes et fonctionnelles dans l’organe concerné. Dans le cas des RP à un stade avancé, où les photorécepteurs et les cellules de l’EP ont dégénéré, la supplémentation n’aura pas d’effet. L’optogénétique est une approche de compensation dont l’objectif est de faire exprimer par d’autres cellules rétiniennes (cellules bipolaires, cellules ganglionnaires, cônes dormants) des protéines photosensibles à même de restaurer la transduction du signal lumineux en signal électrique. À ce jour, la preuve du concept a été obtenue chez l’animal, et deux études de phase I/II chez l’homme sont en cours [2]. Une autre approche compensatrice consiste à reprogrammer des cellules gliales de la rétine afin de stimuler la régénération rétinienne [3].
L’œil, un organe cible idéal pour la thérapie génique
De petite taille, l’œil peut être traité par de fortes concentrations de vecteurs délivrées dans un petit volume. Clos et isolé du reste de l’organisme par la barrière hémato-rétinienne, l’œil est un organe immuno-privilégié, ce qui permet de limiter la dissémination du vecteur et la réaction immunitaire qu’il pourrait induire. Constituée de cellules post-mitotiques (i.e. ne se divisant pas), la rétine permet une expression du gène sur le long terme sans risque d’intégration accidentelle mutagène en cas de division cellulaire [4] (→).
(→) Voir la Synthèse de A. Rossi et A. Salvetti, m/s n° 2, février 2016, page 167
La transparence des milieux qui l’entourent rend la rétine accessible à de multiples examens fonctionnels et d’imagerie non invasive, et permet de suivre la dissémination et l’activité du gène transduit. L’atteinte bilatérale et relativement symétrique des RP permet de traiter un seul œil et de le comparer à l’autre œil, qui sert alors de contrôle. La rétine est facilement accessible chirurgicalement par voie intra-vitréenne ou sous-rétinienne (Figure 2). L’injection sous-rétinienne par voie de vitrectomie6 est une procédure chirurgicale à risque. Elle est cependant préférée actuellement à la voie intra-vitréenne car la transduction du gène thérapeutique y est beaucoup plus efficace [5]. Si l’apport du gène thérapeutique peut s’effectuer ex vivo, (des cellules du patient sont prélevées, modifiées in vitro, puis réinjectées dans l’organe cible), la stratégie in vivo (injection du gène directement dans l’organe cible) est privilégiée en ophtalmologie.
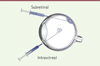 |
Figure 2. Voies d’injection du vecteur : sous-rétinienne ou intra-vitréenne. |
Le vecteur idéal
Pour atteindre la cellule cible, le gène thérapeutique nécessite un véhicule : le vecteur. Les vecteurs non viraux synthétiques sont faciles à produire. Ils peuvent transporter des séquences d’ADN de grande taille et présentent peu de risques de pathogénicité. Malheureusement, leur efficacité in vivo reste faible [6] (→).
(→) Voir le Dossier technique de H. Khabou et D. Dalkara, m/s n° 5, mai 2015, page 529
Les vecteurs viraux sont par contre beaucoup plus efficaces pour acheminer l’ADN dans le noyau de cellules hôtes. Pour être efficace et sûr, le « bon vecteur » nécessite d’être non pathogène, non réplicatif, non immunogène et non intégratif, pour éviter le risque de mutagenèse. Il doit cibler spécifiquement le type cellulaire désiré et permettre une expression du gène sur le long cours. Pour obtenir le profil souhaité en fonction de la maladie et de la cellule cible, il est possible de faire varier de multiples paramètres : la famille du virus utilisé (herpès, adénovirus, lentivirus), son sérotype, sa capside, le promoteur du gène thérapeutique qu’il transporte. Actuellement les virus adéno-associés (AAV) sont les vecteurs les plus utilisés pour le traitement des maladies rétiniennes. Leur principal défaut est la taille relativement petite de la cassette d’expression qu’ils sont capables de transporter et qui ne permet de vectoriser qu’un gène de taille restreinte. Les lentivirus d’origine équine leur sont parfois préférés, du fait de leur capacité de transport plus importante [6]. D’autres facteurs entrent également en compte pour optimiser le transfert du gène : le volume de liquide injecté, la concentration virale, la voie d’injection, le site d’injection (rétine saine ou dégénérée), le stade évolutif de la maladie et la gestion de la fenêtre thérapeutique.
Intérêts et limites des modèles animaux
L’utilisation de modèles animaux est un prérequis, en fonction de leur disponibilité. Des modèles animaux, sauvages ou mutés, sont disponibles pour de nombreuses mutations responsables de RP. Les rats et les souris sont très utilisés en première intention, pour des raisons pratiques et financières. Malheureusement, le faible ratio cônes/bâtonnets, l’absence de macula et des différences dans la transduction limitent la pertinence de ces modèles. Le recours à des grands animaux, tels que le chien et surtout les primates non humains, est nécessaire pour confirmer l’efficacité et la sécurité du traitement avant d’envisager des essais cliniques chez l’homme.
Les données actuelles
Les études cliniques actuelles sont le plus souvent fondées sur une approche (supplémentation et/ou inactivation) qui dépend du gène en cause. Nous présentons ici les résultats des études liées au gène RPE65, seul gène pour lequel un médicament est disponible. Nous citerons ensuite les avancées réalisées pour d’autres gènes (Tableau I).
Avancées actuelles des recherches en thérapie génique pour les rétinites pigmentaires, en fonction du gène causal. Publications ou essais cliniques de moins de 5 ans ou dernière etude publiée dans le domaine. ACL : amaurose congénitale de Leber ; IV : injection intra-vitréenne ; RP : rétinite pigmentaire ; SR : injection sous-rétinienne. * : réalisé chez le rat ou autres petits animaux.
Amaurose congénitale de Leber et RPE65
Le gène RPE65 (retinal pigment epithelium-specific 65) code une rétinoïde isomérohydrolase de 65 kDa. Exprimée dans les cellules de l’EP, elle est indispensable au cycle de la transduction du signal visuel en régénérant le 11-cis rétinal après exposition lumineuse. La mutation bi-allélique de ce gène est responsable de 6 à 16 % des ACL, mais aussi de certaines EOSRD et RP de l’adulte [7]. Cette forme génétique est un bon candidat car, malgré une baisse sévère de la vision dès le jeune âge, les cellules rétiniennes sont encore relativement préservées [8]. Les études précliniques utilisant l’injection sous-rétinienne du gène thérapeutique RPE65 dans un vecteur AAV ont été réalisées chez la souris, puis dans un modèle de chien Briard dépourvu d’expression du gène (RPE65 -/-). Ces études ont démontré l’amélioration des réponses rétiniennes à l’électrorétinogramme et de la fonction visuelle grâce à des tests comportementaux [9-12]. Plusieurs études de phase I/II ont établi la sécurité de la transduction du gène RPE65 thérapeutique et ont révélé une amélioration visuelle chez les patients [13-16]. En 2017, Russel et al. ont publié les résultats d’une étude de phase III, incluant des patients présentant une mutation bi-allélique du gène RPE65 [17]. C’était la première étude randomisée portant sur une thérapie génique en ophtalmologie. Vingt patients ont reçu une injection sous-rétinienne bilatérale de 0,3 ml d’un vecteur AAV2 contenant le gène thérapeutique RPE65 (voretigene neparvovec, ou Luxturna®). Ces 20 patients ont présenté une amélioration modérée mais significative du test de mobilité (critère de jugement principal) par rapport aux neuf patients témoins. Cette amélioration a été observée dès le trentième jour après l’injection dans le deuxième œil et est restée stable pendant un suivi de un an. À un an, 65 % des patients injectés ont réussi le test de mobilité à la plus faible luminosité (1 Lux), contre aucun dans le groupe témoin. Suite à cette étude, la food and drug administration (FDA) et la European medicines agency (EMA) ont donné leur accord pour la commercialisation du Luxturna® (Spark Therapeutics Inc.). Il est actuellement commercialisé aux États-Unis, au prix de 850 000 dollars (environ 765 000 euros) pour les deux yeux. En France, ce produit est pris en charge par l’Assurance maladie et plusieurs patients ont déjà bénéficié de ce traitement dans des centres de référence, notamment au Centre hospitalier national des Quinze-Vingts (CHNO XV-XX) et au Centre hospitalier universitaire de Nantes.
Profil d’efficacité et de sécurité
L’ensemble des études précliniques et cliniques ont montré la bonne tolérance de l’administration sous-rétinienne de vecteurs AAV. Néanmoins, certains points méritent d’être soulignés. Le risque de mutagenèse par insertion accidentelle du gène thérapeutique, avec formation de tumeur, a été documenté pour l’AAV dans le foie de souris nouveau-née [4]. Il semble cependant limité dans la rétine du fait de l’absence de division cellulaire au sein de ce tissu. Une toxicité du vecteur est également possible. Elle dépend de la dose utilisée et du promoteur qui a été choisi our l’expression du gène thérapeutique [18]. Injecté par voie sous-rétinienne chez le rat et le chien, le vecteur a pu être retrouvé dans le nerf optique et les voies visuelles, mais pas dans d’autres organes [19]. La dose de vecteur injectée doit être limitée au minimum nécessaire. Une attention particulière doit également être portée sur les résultats visuels à long terme. En effet, certaines études ont révélé une perte d’efficacité plusieurs années après injection [20, 21].
Technique chirurgicale
Actuellement, l’injection sous-rétinienne est préférée à l’injection intra-vitréenne, car elle met le vecteur injecté directement au contact des cellules cibles, ce qui augmente l’efficacité de vectorisation et diminue les réactions immunitaires et l’exposition inutile d’autres tissus oculaires. Malheureusement, cette technique est invasive. Elle nécessite la réalisation d’une vitrectomie par un chirurgien expérimenté et présente de potentiels effets indésirables : uvéite, cataracte, déchirure rétinienne, décollement de rétine, endophtalmie, pli maculaire, trou maculaire [17, 22]. De nouvelles recherches sont donc nécessaires pour améliorer l’efficacité de la voie intra-vitréenne, la sécurité de l’injection sous-rétinienne, ou pour développer une voie supra-choroïdienne7 [23].
Perspectives
Les autres thérapies géniques de supplémentation
Plusieurs études cliniques de phase I/II évaluent actuellement d’autres traitements de supplémentation ciblant les gènes RPE65, PDE6b (phosphodiestérase 6b), RLPB1 (retinalaldehyde-binding protein 1) et MERTK (myeloid-epithelial-reproductive tyrosine kinase) (Tableau I). Pour la RP liée à l’X (due à des altérations du gène RPGR [retinitis pigmentosa GTPase regulator]), deux études de phase I/II et une de phase I/II/III sont en cours.
L’optogénétique
L’optogénétique consiste à faire exprimer par des cellules rétiniennes des photopigments capables de restaurer la transduction du signal lumineux en signal électrique. Cette méthode présente l’avantage d’être indépendante de la mutation causale, et de la fonctionnalité des photorécepteurs et des cellules de l’EP. Elle serait utilisable à des stades plus avancés d’atrophie rétinienne que pour les stratégies conventionnelles, tant que les cellules bipolaires et/ou ganglionnaires sont fonctionnelles. À ce jour, la preuve de concept a été réalisée chez l’animal et deux études de phase I/II chez l’homme sont prévues8 [2].
Édition de gène : l’apport du système CRISPR/Cas9
Le système CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9) est une découverte majeure du XXIe siècle. Il permet de modifier des séquences nucléotidiques. D’autres techniques d’édition de gène sont également disponibles : l’utilisation de zinc finger nucleases (ZFN), les transcription activator-like effector nucleases (TALEN), ou les méganucléases. Mais le système CRISPR/Cas9 présente plusieurs avantages par rapport à ces autres techniques : il est plus rapide à modéliser et à produire, plus efficace et plus spécifique, il permet aussi l’édition simultanée de plusieurs gènes [24]. Des modèles animaux de RP ont ainsi été développés grâce à ce système. Chez le rat, il permet d’inactiver, par une injection sous-rétinienne unique, l’expression du gène RHO qui code la rhodopsine9, dont l’altération est responsable d’une forme de RP autosomique dominante [25]. Aucune étude n’a encore exploré cette voie en ophtalmologie. La principale limite du système CRISPR/Cas9 est une possible coupure double brin de l’ADN sur des sites non désirés, prévisibles in silico ou non. Ces ciblages off-targets présentent un risque mutagène qui reste difficile à maîtriser et qui mérite une surveillance et des précautions particulières.
Induction de cellules pluripotentes in vitro
Le développement in vitro de lignées de cellules pluripotentes hiPSC (human induced-pluripotent stem cells) à partir de cellules de patient, offre de nouvelles perspectives [26]. Il est désormais possible de dédifférencier des cellules somatiques d’un patient en cellules pluripotentes immortalisées, puis de les redifférencier en cellules de l’EP ou, plus difficilement, en photorécepteurs. Des modèles de rétinites pigmentaires sont actuellement développés. Ils présentent de nombreux avantages. Ils permettent de mieux comprendre la pathogenèse de la maladie et de tester de nouvelles approches thérapeutiques pharmacologiques ou de thérapie génique. Combinée à l’utilisation du système CRISPR/Cas9, l’induction de cellules pluripotentes ouvre de nouveaux horizons : cela permettrait notamment de diminuer significativement le recours aux modèles animaux. Cette technique permet, enfin, d’envisager de développer une thérapie génique ex vivo, en greffant des cellules autologues génétiquement modifiées in vitro.
Aspects économiques et éthiques
Pour trouver sa place dans le système de soin, la thérapie génique doit d’abord trouver son modèle économique. Ce secteur est particulièrement concerné par la loi d’Eroom10 qui prédit que le développement des médicaments sera de plus en plus lent et coûteux. En effet les coûts de développement et de production des thérapies géniques sont très élevés et concernent, pour chaque vecteur, uniquement un gène, voire une mutation particulière d’un gène, et donc peu de patients. On estime ainsi entre 100 et 200 le nombre de patients atteints (en France) de dégénérescence rétinienne liée à la mutation bi-allélique du gène RPE65 et seule une partie d’entre eux conserve suffisamment de rétine fonctionnelle pour que l’injection de Luxturna® leur soit profitable. Alors faut-il rembourser ce produit ? Oui sans doute, car son efficacité, même si elle est modeste, a été démontrée et améliore la qualité de vie des patients, avec une tolérance acceptable. Un signal encourageant fort est donc donné à la communauté des patients, aux chercheurs, aux donateurs privés et aux investisseurs ; mais à l’opposé, on pourrait contester ce « oui » en raison du faible recul que l’on a sur la balance bénéfice/risque et sur le ratio efficacité/coût qui pourrait s’avérer insuffisant [27]. À noter que les patients qui auront profité d’une injection du produit ne pourront plus prétendre à être inclus dans un éventuel essai futur.
Quel prix la solidarité nationale est-elle prête à supporter ? En janvier 2019, le collège de la Haute autorité de santé (HAS) a décidé de mettre en place une évaluation économique et de santé publique du Luxturna®. Il est donc susceptible de connaître le même sort que le Glybera® 11, première thérapie génique autorisée en Europe pour le traitement de déficit familial en lipoprotéine lipase (qui a reçu une autorisation de mise sur le marché [AMM] en 2012). Le Glybera® n’avait cependant pas obtenu de remboursement, en raison d’un intérêt clinique insuffisant. Pour rendre ce modèle viable, il est nécessaire de modifier profondément les procédés de développement et de fabrication afin de diminuer le coût de fabrication du produit, de favoriser des stratégies qui permettraient de rendre les produits confectionnés indépendants de la mutation responsable de la maladie ou d’obtenir des résultats cliniques qui soient très significatifs [59] (→).
(→) Voir le Repères de A. Fischer et al., m/s n° 4, avril 2020, page 389
Une question éthique, est celle du recrutement des patients pour les études cliniques. Le bénéfice attendu des produits est théoriquement bien supérieur chez l’enfant, chez lesquels la dégénérescence rétinienne est moins avancée. Limiter les essais aux adultes pour protéger du risque les enfants est légitime, mais cette option pourrait conduire à des conclusions erronées quant à l’efficacité du traitement, uniquement évaluée chez l’adulte et a priori plus grande chez l’enfant.
Conclusion
La thérapie génique est une approche thérapeutique révolutionnaire en plein essor, dont l’efficacité est désormais établie. Bien au-delà du « simple médicament » qui apporte un gène manquant, de multiples stratégies thérapeutiques sont en cours de développement et permettront de s’adapter au stade de la maladie, qui est évolutive. Le diagnostic et le génotypage le plus précocement possible des patients sont une priorité : ils permettent de proposer des thérapies de supplémentation, d’édition ou d’inactivation du gène muté pour empêcher la dégénérescence rétinienne. Cette notion de « fenêtre thérapeutique » est centrale. Elle est bien illustrée par l’intitulé de l’AMM du Luxturna®, qui réserve le traitement aux patients « possédant suffisamment de cellules viables ». En cas de perte cellulaire avancée, l’optogénétique et la reprogrammation cellulaire sont des modalités prometteuses indépendantes du gène causal. Les perspectives de recherche sont larges et passionnantes, mais il reste de nombreux défis technologiques, économiques et éthiques à surmonter avant que la thérapie génique s’installe dans la pratique médicale.
Liens d’intérêt
Michel Weber est consultant pour Novartis. Michel Weber et Guylène Le Meur sont actionnaires fondateurs de Horama.
Le cycle de la vision est régi par la photoisomérisation du rétinal. Lorsque le 11-cis rétinal absorbe un photon, il passe de l’état 11-cis à l’état tout-trans. Cette isomérisation est à l’origine de l’influx nerveux par phototransduction. Le 11-cis rétinal est ensuite régénéré par voie enzymatique.
https://sph.uth.edu/retnet/
Diminution de la vision en condition d’éclairage faible.
Le nystagmus est un mouvement d’oscillation involontaire et saccadé du globe oculaire causé par une perturbation de la coordination des muscles de l’oeil.
Perte totale de la vue, sans lésion décelable.
Retrait du vitré par voie chirurgicale. Le vitré est la substance qui remplit la cavité oculaire, en arrière du cristallin.
L’espace suprachoroïdien est un espace virtuel situé entre la face externe de la choroïde (la membrane qui tapisse l’intérieur du globe oculaire) et la face interne de la sclère (enveloppe fibreuse externe du globe oculaire).
ClinicalTrials.gov NCT02556736 et ClinicalTrials.gov NCT03326336.
Pigment photosensible des bâtonnets.
La « loi d’Eroom » prédit que chaque découverte d’un nouveau traitement médical prend plus de temps et coûte plus cher que les précédentes, en dépit des progrès technologiques.
Utilisé dans le traitement des adultes présentant un déficit familial en lipoprotéine lipase (LPL) et souffrant de crises de pancréatites sévères ou multiples malgré un régime pauvre en lipides.
Références
- Verbakel SK, van Huet RAC, Boon CJF, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res 2018 ; 66 : 157–186. [Google Scholar]
- Simunovic MP, Shen W, Lin JY, et al. Optogenetic approaches to vision restoration. Exp Eye Res 2019 ; 178 : 15–26. [Google Scholar]
- Jorstad NL, Wilken MS, Grimes WN, et al. Stimulation of functional neuronal regeneration from Müller glia in adult mice. Nature 2017 ; 548 : 103–107. [PubMed] [Google Scholar]
- Rossi A, Salvetti A. Intégration des vecteurs AAV et mutagenèse insertionnelle. Med Sci (Paris) 2016 ; 32 : 167–174. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Seitz IP, Michalakis S, Wilhelm B, et al. Superior retinal gene transfer and biodistribution profile of subretinal versus intravitreal delivery of AAV8 in nonhuman primates. Invest Ophthalmol Vis Sci 2017 ; 58 : 5792–5801. [Google Scholar]
- Khabou H, Dalkara D. La conception de vecteurs adaptés à la thérapie génique oculaire. Med Sci. (Paris) 2015 ; 31 : 529–537. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Miraldi Utz V, Coussa RG, Antaki F, et al. Gene therapy for RPE65-related retinal disease. Ophthalmic Genet 2018; 39 : 671–7. [Google Scholar]
- Chung DC, Traboulsi EI. Leber congenital amaurosis: clinical correlations with genotypes, gene therapy trials update, and future directions. J Am Assoc Pediatr Ophthalmol Strabismus 2009 ; 13 : 587–592. [Google Scholar]
- Acland GM, Aguirre GD, Bennett J, et al. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther 2005 ; 12 : 1072–1082. [Google Scholar]
- Bennicelli J, Wright JF, Komaromy A, et al. Reversal of blindness in animal models of Leber congenital amaurosis using optimized AAV2-mediated gene transfer. Mol Ther 2008 ; 16 : 458–465. [Google Scholar]
- Le Meur G, Stieger K, Smith AJ, et al. Restoration of vision in RPE65-deficient Briard dogs using an AAV serotype 4 vector that specifically targets the retinal pigmented epithelium. Gene Ther 2007 ; 14 : 292–303. [Google Scholar]
- Jacobson SG, Acland GM, Aguirre GD, et al. Safety of recombinant adeno-associated virus type 2-RPE65 vector delivered by ocular subretinal injection. Mol Ther 2006 ; 13 : 1074–1084. [Google Scholar]
- Jacobson SG, Cideciyan AV, Ratnakaram R, et al. Gene therapy for Leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol 2012 ; 130 : 9–24. [Google Scholar]
- Le Meur G, Lebranchu P, Billaud F, et al. Safety and long-term efficacy of AAV4 gene therapy in patients with RPE65 Leber congenital amaurosis. Mol Ther 2018 ; 26 : 256–268. [Google Scholar]
- Bennett J, Wellman J, Marshall KA, et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: a follow-on phase 1 trial. Lancet 2016 ; 388 : 661–672. [Google Scholar]
- Bainbridge JWB, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med 2008 ; 358 : 2231–2239. [Google Scholar]
- Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 2017 ; 390 : 849–860. [Google Scholar]
- Xiong W, Wu DM, Xue Y, et al. AAV cis-regulatory sequences are correlated with ocular toxicity. Proc Natl Acad Sci USA 2019 ; 116 : 5785–5794. [Google Scholar]
- Provost N, Le Meur G, Weber M, et al. Biodistribution of rAAV vectors following intraocular administration: evidence for the presence and persistence of vector DNA in the optic nerve and in the brain. Mol Ther 2005 ; 11 : 275–283. [Google Scholar]
- Jacobson SG, Cideciyan AV, Roman AJ, et al. Improvement and decline in vision with gene therapy in childhood blindness. N Engl J Med 2015 ; 372 : 1920–1926. [Google Scholar]
- Bainbridge JWB, Mehat MS, Sundaram V, et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N Engl J Med 2015 ; 372 : 1887–1897. [Google Scholar]
- Ducloyer JB, Le Meur G, Lebranchu P, et al. Macular fold complicating a subretinal injection of voretigene neparvovec. Ophthalmol Retina 2019; S2468653019306694. [Google Scholar]
- Ding K, Shen J, Hafiz Z, et al. AAV8-vectored suprachoroidal gene transfer produces widespread ocular transgene expression. J Clin Invest 2019 ; 130 : [Google Scholar]
- Cai B, Sun S, Li Z, et al. Application of CRISPR/Cas9 technologies combined with iPSCs in the study and treatment of retinal degenerative diseases. Hum Genet 2018 ; 137 : 679–688. [Google Scholar]
- Bakondi B, Lv W, Lu B, et al. In vivo CRISPR/Cas9 gene editing corrects retinal dystrophy in the S334ter-3 rat model of autosomal dominant retinitis pigmentosa. Mol Ther 2016 ; 24 : 556–563. [Google Scholar]
- Castro AA, Lukovic D, Jendelova P, et al. Concise review: human induced pluripotent stem cell models of retinitis pigmentosa. Stem Cells 2018 ; 36 : 474–481. [Google Scholar]
- Zimmermann M, Lubinga SJ, Banken R, et al. Cost utility of voretigene neparvovec for biallelic RPE65-mediated inherited retinal disease. Value Health 2019 ; 22 : 161–167. [Google Scholar]
- Smalley E.. First AAV gene therapy poised for landmark approval. Nat Biotechnol 2017 ; 35 : 998–999. [Google Scholar]
- Mowat FM, Occelli LM, Bartoe JT, et al. Gene therapy in a large animal model of PDE6A-retinitis pigmentosa. Front Neurosci 2017; 11. [PubMed] [Google Scholar]
- Occelli LM, Schön C, Seeliger MW, et al. Gene supplementation rescues rod function and preserves photoreceptor and retinal morphology in dogs, leading the way toward treating human PDE6A-retinitis pigmentosa. Hum Gene Ther 2017 ; 28 : 1189–1201. [Google Scholar]
- Pichard V, Provost N, Mendes-Madeira A, et al. AAV-mediated gene therapy halts retinal degeneration in PDE6β-deficient dogs. Mol Ther 2016 ; 24 : 867–876. [Google Scholar]
- Feathers KL, Jia L, Perera ND, et al. Development of a gene-therapy vector for RDH12-associated retinal dystrophy. Hum Gene Ther 2019 ; 30 : [Google Scholar]
- Choi VW, Bigelow CE, McGee TL, et al. AAV-mediated RLBP1 gene therapy improves the rate of dark adaptation in Rlbp1 knockout mice. Mol Ther Methods Clin Dev 2015 ; 2 : 15022. [Google Scholar]
- Boye SL, Peterson JJ, Choudhury S, et al. Gene therapy fully restores vision to the all-cone Nrl(-/-) Gucy2e(-/-) mouse model of Leber congenital amaurosis-1. Hum Gene Ther 2015 ; 26 : 575–592. [Google Scholar]
- Zhong H, Eblimit A, Moayedi Y, et al. AAV8(Y733F)-mediated gene therapy in a Spata7 knockout mouse model of Leber congenital amaurosis and retinitis pigmentosa. Gene Ther 2015 ; 22 : 619–627. [Google Scholar]
- Zhang W, Li L, Su Q, et al. Gene therapy using a miniCEP290 fragment delays photoreceptor degeneration in a mouse model of Leber congenital amaurosis. Hum Gene Ther 2018 ; 29 : 42–50. [Google Scholar]
- Mookherjee S, Chen HY, Isgrig K, et al. A CEP290 C-terminal domain complements the mutant CEP290 of Rd16 mice In trans and rescues retinal degeneration. Cell Rep 2018 ; 25 : 611–23 e6. [Google Scholar]
- Garanto A, Chung DC, Duijkers L, et al. In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum Mol Genet 2016 ; 25 : 2552–2563. [PubMed] [Google Scholar]
- Ghazi NG, Abboud EB, Nowilaty SR, et al. Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: results of a phase I trial. Hum Genet 2016 ; 135 : 327–343. [Google Scholar]
- Chekuri A, Sahu B, Chavali VRM, et al. Long-term effects of gene therapy in a novel mouse model of human MFRP-associated retinopathy. Hum Gene Ther 2019 ; 30 : 632–650. [Google Scholar]
- Dinculescu A, Stupay RM, Deng WT, et al. AAV-mediated clarin-1 expression in the mouse retina: implications for USH3A gene therapy. PLoS One 2016 ; 11 : e0148874. [Google Scholar]
- Fischer MD, McClements ME, Martinez-Fernandez de la Camara C, et al. Codon-optimized RPGR improves stability and efficacy of AAV8 gene therapy in two mouse models of X-linked retinitis pigmentosa. Mol Ther 2017 ; 25 : 1854–1865. [Google Scholar]
- Wu Z, Hiriyanna S, Qian H, et al. A long-term efficacy study of gene replacement therapy for RPGR-associated retinal degeneration. Hum Mol Genet 2015 ; 24 : 3956–3970. [Google Scholar]
- Giacalone JC, Andorf JL, Zhang Q, et al. Development of a molecularly stable gene therapy vector for the treatment of RPGR-associated X-linked retinitis pigmentosa. Hum Gene Ther 2019 ; 30 : [Google Scholar]
- Beltran WA, Cideciyan AV, Boye SE, et al. Optimization of retinal gene therapy for X-linked retinitis pigmentosa due to RPGR mutations. Mol Ther 2017 ; 25 : 1866–1880. [Google Scholar]
- Cideciyan AV, Sudharsan R, Dufour VL, et al. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc Natl Acad Sci USA 2018 ; 115 : E8547–E8556. [Google Scholar]
- Botta S, Marrocco E, de Prisco N, et al. Rhodopsin targeted transcriptional silencing by DNA-binding. eLife 2016; 5 : e12242. [Google Scholar]
- Botta S, de Prisco N, Marrocco E, et al. Targeting and silencing of rhodopsin by ectopic expression of the transcription factor KLF15. JCI Insight 2017; 2. pii: 96560. [Google Scholar]
- McCullough KT, Boye SL, Fajardo D, et al. Somatic gene editing of GUCY2D by AAV-CRISPR/Cas9 alters retinal structure and function in mouse and macaque. Hum Gene Ther 2019 ; 30 : 571–589. [Google Scholar]
- Millington-Ward S, Chadderton N, O’Reilly M, et al. Suppression and replacement gene therapy for autosomal dominant disease in a murine model of dominant retinitis pigmentosa. Mol Ther 2011 ; 19 : 642–649. [Google Scholar]
- Roddy GW, Yasumura D, Matthes MT, et al. Long-term photoreceptor rescue in two rodent models of retinitis pigmentosa by adeno-associated virus delivery of Stanniocalcin-1. Exp Eye Res 2017 ; 165 : 175–181. [Google Scholar]
- Byrne LC, Dalkara D, Luna G, et al. Viral-mediated RdCVF and RdCVFL expression protects cone and rod photoreceptors in retinal degeneration. J Clin Invest 2015 ; 125 : 105–116. [Google Scholar]
- Yao K, Qiu S, Wang YV, et al. Restoration of vision after de novo genesis of rod photoreceptors in mammalian retinas. Nature 2018 ; 560 : 484–488. [PubMed] [Google Scholar]
- Jorstad NL, Wilken MS, Grimes WN, et al. Stimulation of functional neuronal regeneration from Müller glia in adult mice. Nature 2017 ; 548 : 103–107. [PubMed] [Google Scholar]
- Cronin T, Vandenberghe LH, Hantz P, et al. Efficient transduction and optogenetic stimulation of retinal bipolar cells by a synthetic adeno-associated virus capsid and promoter. EMBO Mol Med 2014 ; 6 : 1175–1190. [Google Scholar]
- Sengupta A, Chaffiol A, Macé E, et al. Red-shifted channelrhodopsin stimulation restores light responses in blind mice, macaque retina, and human retina. EMBO Mol Med 2016 ; 8 : 1248–1264. [Google Scholar]
- Cheong SK, Strazzeri JM, Williams DR, et al. All-optical recording and stimulation of retinal neurons in vivo in retinal degeneration mice. PLoS One 2018 ; 13 : e0194947. [Google Scholar]
- Khabou H, Garita-Hernandez M, Chaffiol A, et al. Noninvasive gene delivery to foveal cones for vision restoration. JCI Insight 2018; 3. pii: 96029. [Google Scholar]
- Fischer A, Dewatripant M, Goldman M. L’innovation thérapeutique, à quel prix ? Med Sci (Paris) 2020; 36 : 389–93. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des tableaux
Avancées actuelles des recherches en thérapie génique pour les rétinites pigmentaires, en fonction du gène causal. Publications ou essais cliniques de moins de 5 ans ou dernière etude publiée dans le domaine. ACL : amaurose congénitale de Leber ; IV : injection intra-vitréenne ; RP : rétinite pigmentaire ; SR : injection sous-rétinienne. * : réalisé chez le rat ou autres petits animaux.
Liste des figures
|
Figure 1. Structure de la rétine en microscopie confocale. RPE : épithélium pigmentaire ; POS : segment externe des photorécepteurs ; PR : corps cellulaires des photorécepteurs ; HC : cellules horizontales ; BP : cellules bipolaires ; AC : cellules amacrines ; GC : cellules ganglionnaires. |
| Dans le texte | |
|
Figure 2. Voies d’injection du vecteur : sous-rétinienne ou intra-vitréenne. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.