")
")
| Issue |
Med Sci (Paris)
Volume 34, Number 3, Mars 2018
|
|
|---|---|---|
| Page(s) | 247 - 252 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20183403013 | |
| Published online | 16 March 2018 | |
Biais de sexe dans l’asthme allergique
Androgènes et cellules lymphoïdes innées de groupe 2
Sex-bias in allergic asthma: androgens and group 2 innate lymphoid cells
Centre de physiopathologie de Toulouse Purpan (CPTP), Université de Toulouse, Inserm, CNRS, UPS, 31300 Toulouse, France
*
jean-charles.guery@inserm.fr
L’asthme allergique est une maladie inflammatoire chronique caractérisée par une hyperréactivité bronchique. Il touche plus de 10 % de la population et débute souvent dans l’enfance. Il existe des disparités sexuelles dans la prévalence et la sévérité de l’asthme. La maladie est en effet plus fréquente chez les jeunes garçons, mais cette tendance s’inverse à la puberté suggérant un rôle régulateur des hormones sexuelles. Dans cette synthèse, nous résumons les connaissances actuelles sur le rôle des hormones sexuelles dans l’inflammation allergique, en soulignant l’impact des androgènes sur le développement et la fonction des cellules lymphoïdes innées du groupe 2 (ILC2), acteurs critiques des réponses allergiques.
Abstract
Allergic asthma is a chronic pulmonary inflammatory disease initiated by exposure to normally harmless allergens and marked by bronchial hyperreactivity. It affects more than 300 million people worldwide. Asthma often starts in childhood. Epidemiological studies show that there are sexual disparities in the prevalence and severity of asthma. Before the age of 10, the disease is more common in boys. This tendency reverses at puberty suggesting a regulating role of the sex hormones. In this synthesis, we summarize current knowledge on the role of sex hormones in allergic inflammation, with a particular focus on the impact of androgens on the development and function of recently introduced group 2 innate lymphoid cell subsets (ILC2) as critical actors in the initiation of allergic responses.♢
© 2018 médecine/sciences – Inserm
L’asthme allergique est une maladie inflammatoire chronique caractérisée par une hyperréactivité bronchique. Il touche plus de 10 % de la population et débute souvent dans l’enfance. Il existe des disparités sexuelles dans la prévalence et la sévérité de l’asthme. La maladie est en effet plus fréquente chez les jeunes garçons, mais cette tendance s’inverse à la puberté suggérant un rôle régulateur des hormones sexuelles. Dans cette synthèse, nous résumons les connaissances actuelles sur le rôle des hormones sexuelles dans l’inflammation allergique, en soulignant l’impact des androgènes sur le développement et la fonction des cellules lymphoïdes innées du groupe 2 (ILC2), acteurs critiques des réponses allergiques.
Les différences sexuelles dans la prévalence et la sévérité de l’asthme sont bien décrites [1-3]. Cependant, les mécanismes à l’origine de ce biais sexuel ne sont pas définis. Il pourrait reposer principalement sur des différences inhérentes au sexe dans le développement du tissu pulmonaire, son anatomie ou sa physiologie, caractéristiques pouvant être influencées ou non par les hormones sexuelles stéroïdiennes [4]. En parallèle, le microbiote intestinal est apparu comme un acteur clé de la susceptibilité à l’asthme [5] et des études portant sur les maladies autoimmunes suggèrent qu’il pourrait être modifié par les hormones sexuelles au moment de la puberté [6, 7]. Une action directe des hormones sexuelles stéroïdiennes sur des populations de cellules immunitaires particulières impliquées dans l’asthme a par ailleurs été suggérée [8, 9].
Nous ferons dans cette revue, le point sur les connaissances actuelles concernant le rôle des hormones sexuelles dans l’immuno-régulation de l’asthme allergique. Nous discuterons en particulier des résultats récents mettant en évidence l’action protectrice des hormones sexuelles mâles, les androgènes, dans les réponses immunitaires de type 2 à travers la régulation négative des cellules lymphoïdes innées du groupe 2 (ILC2 pour group 2 innate lymphoid cells).
Physiopathologie de l’asthme allergique
L’asthme allergique est une maladie à médiation immunitaire « majoritairement » de type 21 induite par des allergènes, normalement inoffensifs. Chez des sujets génétiquement prédisposés, ces allergènes déclenchent une activation de la réponse immunitaire et une inflammation chronique des voies aériennes supérieures [10]. Comme c’est le cas pour de nombreuses maladies allergiques, l’incidence, la prévalence et la gravité de l’asthme sont différentes entre les hommes et les femmes [1-3]. Le contact de l’allergène, qui présente souvent une activité enzymatique avec les cellules épithéliales pulmonaires provoque des dommages cellulaires qui conduisent à l’activation des cellules dendritiques présentes dans les muqueuses (DC) (Figure 1). Ayant capté l’allergène, les DC migrent vers les ganglions lymphatiques drainants où elles activent des lymphocytes T CD4 naïfs spécifiques de cette antigène et favorisent leur différenciation en lymphocytes de type Th2 (T helper de type 2) qui expriment le facteur de transcription GATA-3 [10]. Les lymphocytes Th2, en sécrétant les IL(interleukine)-4, 5 et 13 contribuent à la pathologie [10]. L’IL-4 amplifie la réponse de type Th2 et contribue, en synergie avec l’IL-13, à la production d’immunoglobulines d’isotype E (IgE) par les lymphocytes B. Ces IgE, en se fixant sur leurs récepteurs de haute affinité (FcɛRI ou récepteur de la région Fc des IgE de type I) exprimés à la surface des mastocytes et des basophiles dans les tissus [10], induisent la libération des médiateurs inflammatoires lorsque les allergènes sont reconnus et captés. L’IL-5 est à l’origine de la différenciation et du recrutement d’éosinophiles, à partir de progéniteurs dans la moelle osseuse. Leur présence dans les tissus, associée à celle d’IgE et aux réponses des mastocytes, est caractéristique de l’asthme allergique. L’IL-13 joue un rôle clé dans la pathologie allergique. Elle agit en effet sur de nombreuses cibles pulmonaires. Elle favorise la production, par l’épithélium bronchique, de médiateurs dont l’éotaxine qui favorise le recrutement des éosinophiles, provoque l’hyperplasie des cellules caliciformes de l’épithélium induisant une production accrue de mucus. Elle agit aussi sur le muscle lisse en augmentant la signalisation calcique et en induisant l’hyper-réactivité bronchique. Elle favorise enfin la production de TGF-β (transforming growth factor beta) par les macrophages, contribuant au remodelage et à la fibrose bronchique. Différents acteurs cellulaires comme les mastocytes, les basophiles et les éosinophiles, en produisant de l’IL-5, de l’IL-13 et de l’IL-4, amplifient ainsi la réponse de type 2 [10]. Ce schéma physiopathologique a été dernièrement complété par la découverte récente des ILC2 [11] ( ) qui a conduit à considérer ces cellules comme un acteur majeur de l’induction des réponses adaptatives de type 2 et la mise en place de l’asthme allergique [10, 12].
) qui a conduit à considérer ces cellules comme un acteur majeur de l’induction des réponses adaptatives de type 2 et la mise en place de l’asthme allergique [10, 12].
() Voir la Synthèse de A. Crinier et al., m/s n° 5, mai 2017, page 534
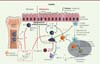 |
Figure 1 Mécanismes d’inhibition par les androgènes de l’inflammation pulmonaire de type 2. Les ILC2 jouent un rôle important dans l’inflammation allergique des voies aériennes. Les allergènes, qui contiennent souvent des protéases, déclenchent la production d’alarmines (TSLP [thymic stromal lymphopoietin], IL[interleukine]-33, IL-25) par les cellules épithéliales. Les cytokines dérivées des cellules épithéliales activent les ILC2 résidentes qui produisent de l’IL-13, une cytokine clé pour la migration des cellules dendritiques (DC) et l’inflammation de type 2. La signalisation androgénique pourrait agir à plusieurs niveaux pour limiter le développement des ILC2 : à l’état basal dans la moelle osseuse sur les progéniteurs des ILC2 (ILC2p) (a) et/ou dans les tissus (b), et lors de la réponse inflammatoire (c). (a) À l’état basal, les androgènes (5a-DHT) limiteraient le développement des ILC2p dans la moelle osseuse en modulant négativement l’expression du récepteur de l’IL-2 [41]. Cette hypothèse n’est cependant pas étayée par des découvertes récentes démontrant que des ILC2 de souris déficientes en CD25 prolifèrent de façon similaire à des ILC2 de souris sauvages, dans un modèle de chimère hématopoïétique, tant à l’état basal que lors d’une infection aiguë par des helminthes [45]. De plus, peu d’ILC2 se développent à partir des progéniteurs de moelle osseuse chez des souris adultes [45]. (b) Le nombre d’ILC2 résidant dans les tissus est plus faible chez les souris mâles [41] et est associé à une diminution de leurs propriétés fonctionnelles ex vivo [42, 46]. Paradoxalement, les ILC2 de souris mâles expriment plus fortement ST-2 (le récepteur de l’IL-33) et KLRG1 (killer cell lectin like receptor G1, un marqueur de maturation). (c) La plupart des différences entre les sexes observées dans le modèle d’inflammation induite par l’IL-33 pourraient être expliquées par une diminution du nombre d’ILC2 résidents à l’état basal [41]. Cependant, un effet direct de la signalisation des androgènes lors de l’expansion des ILC2 ne peut être exclu, comme cela à été récemment suggéré [42]. |
Les ILC2, un acteur important dans l’asthme allergique expérimental
Les ILC2 appartiennent à la famille des cellules lymphoïdes innées dont la nomenclature a été récemment définie [44]. Les ILC2 ne présentent pas de récepteur de l’antigène, mais comme les lymphocytes Th2, ils expriment le facteur de transcription GATA-3. Ils sont activés par des cytokines comme l’IL-33, l’IL-25 et TSLP (thymic stromal lymphopoietin), produites par l’épithélium bronchique à la suite par exemple de leur stimulation par des protéases fréquemment retrouvées dans les allergènes. Les ILC2 sont une source importante d’IL-13 et d’IL-5. Leur activation favorise l’infiltration d’éosinophiles, la sécrétion de mucus et l’hyperréactivité bronchique, mais pas la production d’IgE qui nécessite le développement d’une réponse Th2 adaptative [13]. Des données récentes ont souligné le rôle critique des cytokines de type 2 dérivées des ILC2 dans l’inflammation des poumons induite par certains types d’allergènes, comme les acariens ou les spores du champignon Alternata alternaria. La contribution relative des cytokines produites par les ILC2 et par les lymphocytes Th2 n’est pas encore complètement établie dans l’asthme mais plus de la moitié des cellules productrices de cytokines de type 2 dans les poumons de souris développant un asthme expérimental à l’ovalbumine (OVA) ou aux acariens (HDM) sont des ILC2 [10].
Le développement des ILC2 dépend des facteurs de transcription GATA-3 et RORα [14, 15]. GATA-3 est en effet essentiel au développement des progéniteurs d’ILC2 (les ILC2p) dans la moelle osseuse, et au maintien de la population mature d’ILC2 dans le système périphérique [14]. RORα, un membre de la famille des récepteurs orphelins apparentés au récepteur de l’acide rétinoïque, qui joue un rôle partiellement redondant dans la différenciation des lymphocytes Th17 [16], s’est avéré également critique pour le développement et la fonction des ILC2 [15]. Ainsi, des souris staggered sg/sg, présentant une délétion de RORα au niveau de son domaine de liaison à l’ADN, montrent une réduction drastique du nombre de leurs ILC2 en périphérie et sont incapables de développer une réponse innée efficace contre des parasites intestinaux [15]. Ce modèle murin de déficience en ILC2 a permis de démontrer le rôle clé de ces cellules dans l’initiation des réponses Th2 dans le poumon en réponse à des allergènes de type protéase comme la papaïne [17]. Dans ce modèle, l’IL-13, produite par les ILC2, favorise la migration des DC pulmonaires vers les ganglions lymphatiques drainants où elles initient la différenciation des lymphocytes Th2 [17]. La libération d’IL-33 par les cellules épithéliales qui est nécessaire à l’activation des ILC2 participe à l’inflammation pulmonaire de type 2 [17]. À côté des différentes cytokines épithéliales (IL-33, IL-25 et TSLP apparentée à l’IL-7), les ILC2 détectent également de nombreux signaux environnementaux comme les éicosanoïdes produits par les mastocytes, ou d’autres médiateurs lipidiques comme le leucotriène D4 (LTD4), la prostangladine D2 (PGD2) ou la lipoxine A4 [18-22]. Le LTD4 et la PGD2 agissent de manière synergique avec les cytokines épithéliales pulmonaires. Ils favorisent la production d’IL-13 par les ILC2 [17]. La lipoxine-A4, un médiateur naturel impliqué dans la résolution de l’inflammation, dont l’expression est réduite dans l’asthme sévère, apparaît comme un régulateur négatif de la libération d’IL-13 par les ILC2 chez l’Homme [21].
Deux modèles classiques d’asthme expérimental ont été développés. Le premier (le modèle OVA, pour ovalbumine) est induit par immunisation avec de l’ovalbumine en présence d’alum, les lymphocytes Th2 anti-OVA étant induits par voie systémique. L’inhalation subséquente d’OVA par les animaux immunisés conduit au recrutement des lymphocytes Th2 mémoires dans les poumons qui sont à l’origine de la cascade inflammatoire responsable de la pathologie. Le second modèle est induit par des sensibilisations multiples d’extraits d’acariens par voie aérienne (modèle HDM, pour house dust mice). La réponse immune est alors initiée dans les poumons. Ce modèle est donc plus proche de la pathologie humaine. L’utilisation de ces deux modèles d’asthme chez des souris RORα mutantes staggered sg/sg a permis de révéler des distinctions importantes dans la contribution des ILC2 dans le développement de l’inflammation pulmonaire allergique. Alors que les ILC2 sont nécessaires dans le modèle HDM pour le développement de l’asthme, l’absence d’ILC2 n’a que peu d’impact dans le modèle OVA. Il est à noter que, dans ce dernier modèle, la réponse Th2 est facilitée par l’utilisation de l’adjuvant alum, qui stimule la production d’acide urique, un signal de danger suffisant pour induire une polarisation Th2 par les DC [23]. Les ILC2 joueraient donc un rôle critique lors de l’induction de la réponse Th2 adaptative aux allergènes inhalés. Ils ne seraient cependant pas nécessaires à la stimulation des cellules Th2 mémoires induites par voie systémique [23]. La découverte des ILC2 a donc permis de mieux comprendre les mécanismes mis en jeu dans l’inflammation pulmonaire de type 2 induite par les allergènes inhalés (résumés dans la Figure 1).
Différences sexuelles dans l’asthme allergique
L’asthme fluctue au cours de la vie du patient qui connaît des périodes de rémission et de rechute [3]. Ceci est particulièrement vrai si l’on considère les patients asthmatiques dont la maladie a débuté au cours de l’enfance. Alors que la prévalence de l’asthme est plus élevée chez les garçons que chez les filles avant l’adolescence, cette tendance s’inverse à la puberté non seulement pour l’asthme, mais aussi pour la plupart des troubles allergiques [1, 3]. Bien que cette évolution ait été attribuée à une incidence tardive de l’asthme chez les filles [2, 24], une étude récente met en évidence une fréquence plus élevée de rémissions chez les garçons que chez les filles [25] : entre 10 et 18 ans, le pourcentage de sujets devenant asymptomatiques pour l’asthme est en effet de 39,4 % chez les garçons et de 23,4 % chez les filles [25]. Pendant cette période, l’apparition de l’asthme tend à être plus élevée chez les filles que chez les garçons. Le biais sexuel dans la prévalence de l’asthme pendant l’enfance s’inverse donc à l’adolescence et chez les jeunes adultes [1-3]. La diminution de l’incidence de l’asthme que l’on observe pendant et après la puberté chez les hommes, ainsi que l’augmentation du nombre de rémissions observée chez les hommes, suggèrent ainsi fortement une action protectrice des hormones sexuelles mâles sur le développement de l’asthme [1, 2]. Ces observations sont en adéquation avec le modèle généralement accepté selon lequel les androgènes, qui sont produits à des concentrations plus élevées chez les hommes après la puberté, exerceraient plutôt une action inhibitrice sur la réponse immunitaire [26]. Les mécanismes cellulaires et moléculaires restent cependant très mal compris [26, 27]. Récemment, il a été démontré que la signalisation androgénique modifiait les fonctions des cellules Th1 via l’inhibition de la signalisation du récepteur de l’IL-12 dans les lymphocytes T CD4 [27]. Ce rare exemple d’un effet inhibiteur des androgènes via un effet intrinsèque sur les lymphocytes T pourrait ainsi rendre compte de la plus grande susceptibilité des mâles aux infections virales et au développement de certains cancers [26]. Dans l’asthme, l’inhibition de la différenciation des lymphocytes en lymphocytes de type Th1 par les androgènes devrait favoriser le développement des lymphocytes Th2. Ce mécanisme ne peut donc pas expliquer une possible action immunosuppressive des androgènes sur le développement d’inflammation pulmonaire de type 2.
Effet des hormones sexuelles sur le développement et l’évolution de l’inflammation de type 2
Dans le modèle OVA d’asthme allergique, des biais de réponse entre les sexes ont été rapportés à plusieurs reprises. Dans la plupart des études, les infiltrats d’éosinophiles, la production d’IgE sériques et de cytokines de type 2, l’hyperréactivité bronchique et le remodelage des tissus pulmonaires sont augmentés chez les souris femelles par rapport aux mâles [28-30]. Une susceptibilité accrue des femelles à l’asthme allergique a également été observée dans le modèle HDM [29]. Le rôle protecteur des androgènes dans l’asthme allergique a par ailleurs été suggéré dans une étude qui montrait que la castration de souris mâles abolissait les différences entre les sexes [31]. En faveur de cette hypothèse, l’administration de déhydroépiandrostérone (DHEA), une hormone stéroïde naturelle sécrétée par la glande surrénale et convertie en androgènes ou en œstrogènes, supprime l’infiltration des éosinophiles et l’hyperréactivité bronchique dans le modèle d’asthme induit par l’OVA [32]. Des effets bénéfiques de la castration ont également été décrits chez les souris femelles, dans le modèle d’asthme à l’OVA, sans que le rôle des œstrogènes soit clairement établi [33]. Les œstrogènes agissent via deux récepteurs nucléaires REα et REβ qui régulent l’expression de gènes cibles de manière directe ou indirecte. Les deux isotypes du récepteur sont exprimés dans les poumons [34]. Une déficience en REα dans des souris femelles est associée à une hyperréactivité bronchique dans les conditions basales, sans modifier toutefois l’inflammation de type 2 induite dans le modèle d’asthme à l’OVA [35]. Ces résultats suggèrent que la signalisation œstrogénique régulerait l’activité du muscle lisse préférentiellement à celle des cellules immunocompétentes [35]. La castration, ou la supplémentation en œstrogènes, des souris sauvages n’a pas permis de retrouver le phénotype des souris déficientes pour REα, suggérant un mécanisme d’action indépendant du récepteur et de son ligand [35]. De manière paradoxale, des effets protecteurs de la supplémentation en 17β-estradiol (E2) ont été rapportés dans le modèle d’asthme à l’OVA chez les souris BALB/c ovariectomisées [36]. Dans ce modèle, la supplémentation en E2 inhibe de manière dose-dépendante tous les paramètres cardinaux de l’asthme, comme l’inflammation pulmonaire et l’hyperréactivité bronchique [36]. Ces observations illustrent la complexité des effets des hormones sexuelles administrées de manière pharmacologique, probablement liée à l’expression pléiotropique de leurs récepteurs nucléaires dans de nombreux tissus, incluant le système immunitaire. Ainsi, l’E2 administré de manière pharmacologique a été associé à des effets pro-ou anti-inflammatoires sur l’immunité adaptative par des mécanismes distincts selon les modèles [37, 38]. Le rôle des œstrogènes comme régulateur positif de l’inflammation de type 2 dans l’asthme reste donc très controversé. Bien que nous ne puissions exclure une activité des œstrogènes sur les cellules immunitaires résidant dans les tissus, comme les DC [38] ou les macrophages [39], il est peu probable que ces hormones contribuent de manière majeure aux différences sexuelles dans l’asthme.
Régulation des ILC2 par les androgènes : implication dans l’inflammation de type 2
À l’adolescence, une modification de l’incidence de l’asthme chez les deux sexes se produit. Après la puberté, les femmes deviennent plus susceptibles à l’asthme que les hommes. Les ILC2 jouant un rôle central dans l’induction de l’inflammation des voies respiratoires, l’impact des facteurs dépendants du sexe sur la biologie des ILC2 a été récemment étudié [40-42]. Dans un modèle d’asthme OVA induit chez des souris BALB/c, mâles ou femelles, les ILC2 issues de souris femelles produisent significativement plus d’IL-5 et d’IL-13 que les cellules isolées des souris mâles [40]. Nous avons évalué l’impact du sexe et des hormones sexuelles sur la biologie des ILC2, à l’état basal et en conditions inflammatoires [41]. Les conclusions principales de nos travaux sont résumées dans la Figure 1. En absence d’inflammation, les souris femelles ont un nombre accru d’ILC2 dans les tissus dans lesquels ces cellules se localisent préférentiellement (le tissu adipeux ou associé aux muqueuses). Les souris femelles présentent deux fois plus d’ILC2 que les souris mâles au niveau des poumons. Les ILC2 de mâles et de femelles sont phénotypiquement différentes : l’expression de KLRG1 (killer-cell lectin like receptor G1), un récepteur lectinique exprimé par les cellules NK (natural killer), et du récepteur de l’IL-33 (interleukin 1 receptor-like 1, ST2/IL1RL1) est, par exemple, augmentée à la surface des ILC2 des souris mâles. Ces résultats suggèrent des différences dans le développement des ILC2. En faveur de cette hypothèse, la fréquence des précurseurs d’ILC2 (ILC2p) est plus élevée chez les femelles. Les souris mâles développent donc une inflammation pulmonaire moins sévère que les femelles lorsqu’on leur injecte de l’IL-33 à l’origine de l’expansion et de l’activation de ces cellules. Ce biais dans le nombre des ILC2 dépend non pas des hormones sexuelles féminines mais des hormones mâles, les androgènes. L’ovariectomie et la déficience en REα n’ont en effet pas de conséquences sur le développement des ILC2. En revanche, l’orchidectomie2 ou la déficience en récepteurs des androgènes dans les cellules hématopoïétiques, abolit tous les changements phénotypiques de l’inflammation pulmonaire relayée par l’IL-33. Les précurseurs des ILC2 dans la moelle osseuse (ILC2p) expriment sélectivement le gène Ar codant le récepteur des androgènes, tandis que les gènes codant les récepteurs des œstrogènes Esr-1 (REα) et Esr-2 (REβ) sont peu exprimés. Ar est un gène de signature typique des ILC2 résidant dans les tissus [43]. Des gènes orthologues d’Ar et d’autres gènes de signature ILC2 ont été identifiés chez les vertébrés inférieurs, suggérant que l’expression de ce gène par les ILC2 pourrait avoir été sélectionnée très tôt au cours de l’évolution [44]. Les ILC2p peuvent reconstituer efficacement le compartiment ILC2, par un transfert adoptif. Ils représentent donc un modèle utile pour suivre la différenciation ILC2 et la réponse aux stimulus in vivo et in vitro [14]. Nous avons montré que les androgènes inhibent significativement la maturation des ILC2p en ILC2, et que cet effet peut être levé par l’ajout d’un antagoniste du récepteur des androgènes [41]. Cependant, très peu d’ILC2 se développent à partir de progéniteurs de moelle osseuse chez des souris adultes [45]. Nous pensons donc que la plupart des différences sexuelles que l’on observe dans le modèle inflammatoire induit par l’IL-33 sont préexistantes à l’état inflammatoire, ce qui est en accord avec les différences quantitatives d’ILC2 dans les tissus mâles et femelles, à l’état basal. Nous ne pouvons exclure que les androgènes puissent inhiber également la prolifération des ILC2 localisées dans les tissus. Une étude in vitro récente montre en effet que la production de cytokines par les ILC2 est inhibée par les androgènes [42]. Ces études montrent donc que les androgènes inhibent non seulement le développement des ILC2, mais également leur capacité à produire des cytokines in vitro [40, 42]. Il reste désormais à déterminer si la signalisation androgénique peut contribuer à la régulation négative de la prolifération et des fonctions effectrices des ILC2 in vivo et par quels mécanismes.
Conclusions
Des changements dans la prévalence de nombreux troubles allergiques ont été observés en fonction du sexe, de la petite enfance à l’adolescence. Bien que les œstrogènes aient longtemps été soupçonnés d’être responsables du biais de réponse lié au sexe dans l’asthme, il existe peu de données soutenant un rôle crucial de ces hormones dans l’inflammation de type 2. Des études récentes portant sur l’étude des ILC2 ont en revanche montré un impact majeur des androgènes sur leur fonction [40, 42] et leur développement à l’état basal et lors d’une réponse inflammatoire in vivo [41, 42]. Il a été établi que cette action inhibitrice des androgènes nécessitait l’activation intrinsèque du récepteur des androgènes dans ces cellules [41]. Ces résultats, qui doivent être confirmés chez l’Homme, pourraient expliquer l’inversion du sex-ratio dans la prévalence de l’asthme après la puberté. En ce sens, des données récentes ont montré que le nombre d’ILC2 était augmenté dans le sang des femmes atteintes d’asthme sévère en comparaison aux hommes [42]. Dans l’ensemble, ces travaux révèlent un aspect inattendu de la biologie des ILC2 qui pourrait avoir des implications thérapeutiques, en particulier dans l’asthme et l’allergie. ♢
Remerciements
Ce travail a été soutenu par des subventions de la Fondation pour la recherche médicale (DEQ2000329169) et du Conseil régional Midi-Pyrénées. Nous remercions le Dr Lucette Pelletier pour ses commentaires et suggestions.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Référence
- Carey MA, Card JW, Voltz JW, et al. It’s all about sex: gender, lung development and lung disease. Trends Endocrinol Metab 2007; 18 : 308-13. [CrossRef] [PubMed] [Google Scholar]
- Almqvist C, Worm M, Leynaert B, working group of GALENWPG. Impact of gender on asthma in childhood and adolescence: a GA2LEN review. Allergy 2008; 63 : 47-57. [PubMed] [Google Scholar]
- Holgate ST, Wenzel S, Postma DS, et al. Asthma. Nat Rev Dis Primers 2015; 1 : 15025. [CrossRef] [Google Scholar]
- Townsend EA, Miller VM, Prakash YS. Sex differences and sex steroids in lung health and disease. Endocr Rev 2012; 33 : 1-47. [CrossRef] [PubMed] [Google Scholar]
- Lambrecht BN, Hammad H. The immunology of the allergy epidemic and the hygiene hypothesis. Nat Immunol 2017; 18 : 1076-83. [CrossRef] [PubMed] [Google Scholar]
- Markle JG, Frank DN, Mortin-Toth S, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013; 339 : 1084-8. [Google Scholar]
- Yurkovetskiy L, Burrows M, Khan AA, et al. Gender bias in autoimmunity is influenced by microbiota. Immunity 2013; 39 : 400-12. [CrossRef] [PubMed] [Google Scholar]
- Keselman A, Heller N. Estrogen signaling modulates allergic inflammation and contributes to sex differences in asthma. Front Immunol 2015; 6 : 568. [CrossRef] [PubMed] [Google Scholar]
- Fuseini H, Newcomb DC. Mechanisms driving gender differences in asthma. Curr Allergy Asthma Rep 2017; 17 : 19. [CrossRef] [PubMed] [Google Scholar]
- Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol 2015; 16 : 45-56. [CrossRef] [PubMed] [Google Scholar]
- Crinier A, Viant C, Girard-Madoux M, Vivier E. Les cellules lymphoïdes innées. Med Sci (Paris) 2017; 33 : 534-42. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Neill DR, Wong SH, Bellosi A, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010; 464 : 1367-70. [CrossRef] [PubMed] [Google Scholar]
- Martinez-Gonzalez I, Steer CA, Takei F. Lung ILC2s link innate and adaptive responses in allergic inflammation. Trends Immunol 2015; 36 : 189-95. [CrossRef] [PubMed] [Google Scholar]
- Hoyler T, Klose CS, Souabni A, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity 2012; 37 : 634-48. [CrossRef] [PubMed] [Google Scholar]
- Wong SH, Walker JA, Jolin HE, et al. Transcription factor RORalpha is critical for nuocyte development. Nat Immunol 2012; 13 : 229-36. [CrossRef] [PubMed] [Google Scholar]
- Yang XO, Pappu BP, Nurieva R, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 2008; 28 : 29-39. [CrossRef] [PubMed] [Google Scholar]
- Halim TY, Steer CA, Matha L, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 2014; 40 : 425-35. [CrossRef] [PubMed] [Google Scholar]
- Doherty TA, Khorram N, Lund S, et al. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J Allergy Clin Immunol 2013; 132 : 205-13. [CrossRef] [PubMed] [Google Scholar]
- Xue L, Salimi M, Panse I, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol 2014; 133 : 1184-94. [CrossRef] [PubMed] [Google Scholar]
- Wojno ED, Monticelli LA, Tran SV, et al. The prostaglandin D(2) receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal Immunol 2015; 8 : 1313-23. [Google Scholar]
- von Moltke J, O’Leary CE, Barrett NA, et al. Leukotrienes provide an NFAT-dependent signal that synergizes with IL-33 to activate ILC2s. J Exp Med 2017; 214 : 27-37. [CrossRef] [PubMed] [Google Scholar]
- Barnig C, Cernadas M, Dutile S, et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci TranslMed 2013; 5 : 174ra26. [Google Scholar]
- Gold MJ, Antignano F, Halim TY, et al. Group 2 innate lymphoid cells facilitate sensitization to local, butnot systemic, TH2-inducing allergen exposures. J Allergy Clin Immunol 2014; 133 : 1142-8. [CrossRef] [PubMed] [Google Scholar]
- Nicolai T, Pereszlenyiova-Bliznakova L, Illi S, et al. Longitudinal follow-up of the changing gender ratio in asthma from childhood to adulthood: role of delayed manifestation in girls. Pediatr Allergy Immunol 2003; 14 : 280-3. [CrossRef] [PubMed] [Google Scholar]
- Arshad SH, Raza A, Lau L, et al. Pathophysiological characterization of asthma transitions across adolescence. Respir Res 2014; 15 : 153. [CrossRef] [PubMed] [Google Scholar]
- Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol 2016; 16 : 626-38. [CrossRef] [PubMed] [Google Scholar]
- Kissick HT, Sanda MG, Dunn LK, et al. Androgens alter T-cell immunity by inhibiting T-helper 1 differentiation. Proc Nat! Acad Sci USA 2014; 111 : 9887-92. [CrossRef] [Google Scholar]
- Melgert BN, Postma DS, Kuipers I, et al. Female mice are more susceptible to the development of allergic airway inflammation than male mice. Clin Exp Allergy 2005; 35 : 1496-503. [CrossRef] [PubMed] [Google Scholar]
- Blacquiere MJ, Hylkema MN, Postma DS, et al. Airway inflammation and remodeling in two mouse models of asthma: comparison of males and females. Int Arch Allergy Immunol 2010; 153 : 173-81. [CrossRef] [PubMed] [Google Scholar]
- Takeda M, Tanabe M, Ito W, et al. Gender difference in allergic airway remodelling and immunoglobulin production in mouse model of asthma. Respirology 2013; 18 : 797-806. [CrossRef] [PubMed] [Google Scholar]
- Hayashi T, Adachi Y, Hasegawa K, Morimoto M. Less sensitivity for late airway inflammation in males than females in BALB/c mice. Scand J Immunol 2003; 57 : 562-7. [CrossRef] [PubMed] [Google Scholar]
- Liou CJ, Huang WC. Dehydroepiandrosterone suppresses eosinophil infiltration and airway hyperresponsiveness via modulation of chemokines and Th2 cytokines in ovalbumin-sensitized mice. J Clin Immunol 2011; 31 : 656-65. [CrossRef] [PubMed] [Google Scholar]
- Riffo-Vasquez Y, Ligeiro de Oliveira AP, Page CP, et al. Role of sex hormones in allergic inflammation in mice. Clin Exp Allergy 2007; 37 : 459-70. [CrossRef] [PubMed] [Google Scholar]
- Couse JF, Lindzey J, Grandien K, et al. Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout mouse. Endocrinology 1997; 138 : 4613-21. [CrossRef] [PubMed] [Google Scholar]
- Carey MA, Card JW, Bradbury JA, et al. Spontaneous airway hyperresponsiveness in estrogen receptor-alpha-deficient mice. Am J Respir Crit Care Med 2007; 175 : 126-35. [CrossRef] [PubMed] [Google Scholar]
- Dimitropoulou C, Drakopanagiotakis F, Chatterjee A, et al. Estrogen replacement therapy prevents airway dysfunction in a murine model of allergen-induced asthma. Lung 2009; 187 : 116-27. [CrossRef] [PubMed] [Google Scholar]
- Laffont S, Garnier L, Lelu K, Guery JC. Estrogen-mediated protection of experimental autoimmune encephalomyelitis: Lessons from the dissection of estrogen receptor-signaling in vivo. BiomedJ 2015; 38 : 194-205. [CrossRef] [Google Scholar]
- Laffont S, Seillet C, Guery JC. Estrogen receptor-dependent regulation of dendritic cell development and function. Front Immunol 2017; 8 : 108. [PubMed] [Google Scholar]
- Melgert BN, Oriss TB, Qi Z, et al. Macrophages: regulators of sex differences in asthma? Am J Respir Cell Mol Biol 2010; 42 : 595-603. [CrossRef] [Google Scholar]
- Warren KJ, Sweeter JM, Pavlik JA, et al. Sex differences in activation of lung-related type 2 innate lymphoid cells in experimental asthma. Ann Allergy Asthma Immunol 2017; 118 : 233-34. [CrossRef] [PubMed] [Google Scholar]
- Laffont S, Blanquart E, Savignac M, et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J Exp Med 2017; 214 : 1581-92. [CrossRef] [PubMed] [Google Scholar]
- Cephus J-Y, Stier MT, Fuseini H, et al. Testosterone attenuates group 2 innate lymphoid cell-mediated airway inflammation. Cell Rep 2017; 21 : 2487-99. [CrossRef] [PubMed] [Google Scholar]
- Robinette ML, Fuchs A, Cortez VS, et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol 2015; 16 : 306-17. [CrossRef] [PubMed] [Google Scholar]
- Vivier E, van de Pavert SA, Cooper MD, Belz GT. The evolution of innate lymphoid cells. Nat Immunol 2016; 17 : 790-4. [CrossRef] [PubMed] [Google Scholar]
- Gasteiger G, Fan X, Dikiy S, et al. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 2015; 350 : 981-5. [Google Scholar]
- Warren KJ, Sweeter JM, Pavlik JA, et al. Sex differences in activation of lung-related type 2 innate lymphoid cells in experimental asthma. Ann Allergy Asthma Immunol 2017; 118 : 233-4. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1 Mécanismes d’inhibition par les androgènes de l’inflammation pulmonaire de type 2. Les ILC2 jouent un rôle important dans l’inflammation allergique des voies aériennes. Les allergènes, qui contiennent souvent des protéases, déclenchent la production d’alarmines (TSLP [thymic stromal lymphopoietin], IL[interleukine]-33, IL-25) par les cellules épithéliales. Les cytokines dérivées des cellules épithéliales activent les ILC2 résidentes qui produisent de l’IL-13, une cytokine clé pour la migration des cellules dendritiques (DC) et l’inflammation de type 2. La signalisation androgénique pourrait agir à plusieurs niveaux pour limiter le développement des ILC2 : à l’état basal dans la moelle osseuse sur les progéniteurs des ILC2 (ILC2p) (a) et/ou dans les tissus (b), et lors de la réponse inflammatoire (c). (a) À l’état basal, les androgènes (5a-DHT) limiteraient le développement des ILC2p dans la moelle osseuse en modulant négativement l’expression du récepteur de l’IL-2 [41]. Cette hypothèse n’est cependant pas étayée par des découvertes récentes démontrant que des ILC2 de souris déficientes en CD25 prolifèrent de façon similaire à des ILC2 de souris sauvages, dans un modèle de chimère hématopoïétique, tant à l’état basal que lors d’une infection aiguë par des helminthes [45]. De plus, peu d’ILC2 se développent à partir des progéniteurs de moelle osseuse chez des souris adultes [45]. (b) Le nombre d’ILC2 résidant dans les tissus est plus faible chez les souris mâles [41] et est associé à une diminution de leurs propriétés fonctionnelles ex vivo [42, 46]. Paradoxalement, les ILC2 de souris mâles expriment plus fortement ST-2 (le récepteur de l’IL-33) et KLRG1 (killer cell lectin like receptor G1, un marqueur de maturation). (c) La plupart des différences entre les sexes observées dans le modèle d’inflammation induite par l’IL-33 pourraient être expliquées par une diminution du nombre d’ILC2 résidents à l’état basal [41]. Cependant, un effet direct de la signalisation des androgènes lors de l’expansion des ILC2 ne peut être exclu, comme cela à été récemment suggéré [42]. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.