")
")
| Issue |
Med Sci (Paris)
Volume 33, Number 3, Mars 2017
Autophagie
|
|
|---|---|---|
| Page(s) | 252 - 259 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20173303011 | |
| Published online | 03 April 2017 | |
L’autophagie dans les maladies chroniques du foie
Un ami qui vous veut (presque) toujours du bien !
Autophagy in chronic liver diseases: a friend rather than a foe?
1
Inserm, U1065, C3M, Team 8 “Hepatic complications in obesity”, Nice, France
2
Université Nice Côte d’Azur, Inserm, C3M, Nice, France
3
Institut Cochin, Inserm, U1016, CNRS UMR 8104, université Paris-Descartes, Paris, France
4
Inserm-U1149, CNRS-ERL8252, Centre de recherche sur l’inflammation, Paris, France
5
Sorbonne Paris Cité, Laboratoire d’excellence Inflamex, faculté de médecine, site Xavier Bichat, université Paris Diderot, Paris, France
a
This email address is being protected from spambots. You need JavaScript enabled to view it.
b
This email address is being protected from spambots. You need JavaScript enabled to view it.
c
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
L’autophagie est un processus de dégradation lysosomale qui permet de maintenir l’état énergétique de la cellule en recyclant les protéines et les organites endommagés. Elle joue un rôle majeur dans l’homéostasie hépatique et est dérégulée au cours des maladies chroniques du foie, notamment la maladie alcoolique (ALD) et la stéatopathie non alcoolique (NAFLD). ALD et NAFLD présentent en effet des traits histopathologiques communs allant de la simple stéatose à la stéatohépatite et à la fibrose du foie qui peut, en cas de maintien de l’agression chronique, progresser vers la cirrhose et le carcinome hépatocellulaire. Nous discuterons de l’implication de l’autophagie dans ces différentes phases de progression de la maladie, la régénération et la carcinogenèse hépatiques. En modifiant son angle de vue, c’est-à-dire le type cellulaire étudié, hépatocyte, macrophage ou cellule étoilée, l’histoire est toute différente et les perspectives thérapeutiques en deviennent plus complexes.
Abstract
Within recycling damaged cell components, autophagy maintains cell homeostasis. Thus, it has been anticipated that autophagy would play an essential role in the pathogenesis of chronic liver diseases. Alcoholic liver disease (ALD) and non-alcoholic fatty liver disease (NAFLD) are the most prevalent chronic liver diseases in Western countries, sharing common histopathologic features and a common disease progression. In this review, we discuss the role of autophagy at different stages of NAFLD and ALD as well as in liver regeneration and hepatocarcinogenesis.
© 2017 médecine/sciences – Inserm
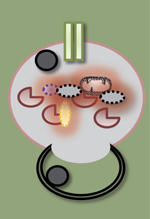
Sous le terme « maladies chroniques du foie » sont regroupées des pathologies d’étiologies très variées, en particulier virale, auto-immune, toxique ou métabolique. Cependant, l’alcool et le surpoids sont à l’origine des maladies les plus fréquentes appelées respectivement « maladie alcoolique du foie » (ALD pour alcoholic liver disease) et « stéatopathie métabolique » (NAFLD pour non-alcoholic fatty liver disease). Leur prévalence atteint en effet 20 à 30 % dans les pays développés. La NAFLD est généralement associée au syndrome métabolique qui regroupe plusieurs des éléments suivants : obésité, diabète de type 2 ou insulino-résistance, dyslipidémie et/ou hypertension artérielle. L’ALD est, quant à elle, consécutive à une consommation chronique excessive d’alcool (> 20 g d’alcool/jour, soit 2 verres de vin chez la femme et > 30 g/jour, soit 3 verres de vin chez l’homme). Ces maladies partageant des caractéristiques communes et une évolution similaire, cette revue abordera le rôle complexe de l’autophagie dans ces deux types d’affection.
NAFLD et ALD : une étiologie différente mais une histopathologie commune…
La stéatose est l’un des traits communs initiateurs de la NAFLD et de l’ALD. Elle se définit par l’accumulation d’acides gras (AG) estérifiés en triglycérides intra-hépatocytaires et peut évoluer vers une stéatohépatite (encore appelée NASH pour non-alcoholic steatohepatitis ou ASH pour alcoholic steatohepatitis) qui associe une souffrance hépatocytaire (ballonisation des hépatocytes, apoptose), une infiltration de cellules inflammatoires et l’activation du processus fibrogénique. Dans 20 % des cas, si rien ne vient interrompre l’agression chronique, comme l’arrêt de l’alcool dans l’ALD, la progression de la fibrose aboutira à une cirrhose en une vingtaine d’années [1, 2]. Par ailleurs, quelle qu’en soit l’origine, la stéatose hépatique induit une diminution des capacités régénératives du foie et les patients qui en sont porteurs présentent un risque accru de développer un hépatocarcinome (Figure 1).
 |
Figure 1. Les maladies métaboliques chroniques du foie : ALD (alcoholic liver disease) et NAFLD (non-alcoholic liver disease) partagent des traits communs. L’obésité et la consommation chronique d’alcool sont responsables d’un spectre de lésions histologiques regroupées sous le terme de « stéatopathies », allant de l’accumulation de gras dans le foie (stéatose) à l’association de la stéatose et de l’inflammation (stéatohépatite) et à l’activation du processus fibrogénique conduisant à la cirrhose et à l’insuffisance hépatique associée à une hypertension portale et une encéphalopathie hépatique. La progression vers ces différentes étapes sont dépendantes non seulement de mécanismes intra- mais également extra-hépatiques, touchant notamment (1) l’intestin avec une dysbiose et une augmentation de la perméabilité intestinale participant à l’inflammation hépatique via les motifs moléculaires associés aux pathogènes (PAMP), dont le lipopolysaccharide (LPS), et (2) le tissu adipeux et son flux accru d’acides gras libres (AGL) et d’adipokines qui participent à l’apoptose hépatocytaire et à l’inflammation hépatique. Ces deux éléments sont essentiels au processus fibrogénique conduisant à la cirrhose. Chacune de ces étapes est associée à un défaut de régénération du foie. |
La stéatose est la conséquence d’une combinaison d’altérations du métabolisme lipidique induites par l’alcool ou l’obésité à savoir : (1) une augmentation de la lipolyse dans le tissu adipeux, qui conduit à la libération d’acides gras non estérifiés acheminés vers le foie ; (2) une augmentation hépatique de la synthèse de novo d’AG ou lipogenèse, combinée à une baisse de la bêta-oxydation mitochondriale des AG ; et enfin (3) une diminution de la libération dans la circulation des triglycérides hépatiques sous forme de VLDL (very low density lipoproteins ou lipoprotéines de très basse densité). De plus, dans l’ALD, certains métabolites de l’alcool, en particulier l’acétaldéhyde, ont des effets directs sur le rapport NAD (nicotinamide adenine dinucleotide)+/NADH induisant des dommages mitochondriaux intra-hépatocytaires.
Les mécanismes qui sous-tendent la transition entre stéatose et NASH sont complexes et encore mal compris (Figure 1). Sont notamment impliqués des métabolites lipotoxiques, un stress oxydatif ainsi qu’une altération des organites cellulaires tels que les mitochondries, les lysosomes et le réticulum endoplasmique (RE) [1, 3]. De plus, les deux types d’affection présentent une activation du système immunitaire inné et adaptatif, induite en partie par la translocation de composés microbiens. Celle-ci est à l’origine d’une réponse inflammatoire participant à la progression de la maladie [4]. Enfin, le tissu adipeux contribue également à la pathogénie de la NAFLD en induisant un état d’inflammation systémique chronique de bas grade et une altération de la sécrétion de cyto/adipokines [5, 6].
… Et un destin commun : fibrose, cancer et défaut de régénération
En cas d’agression chronique persistante, NAFLD et ALD partageront également un destin commun : la constitution d’une fibrose hépatique qui évoluera progressivement vers son point d’orgue, la cirrhose. La fibrose est un déséquilibre entre une production excessive de protéines matricielles par une population hétérogène de myofibroblastes, parmi lesquelles les cellules étoilées (HSC pour hepatic stellate cells) et une insuffisance de remodelage de cette matrice. Dans le foie normal, les HSC sont quiescentes et leur fonction principale est de stocker la vitamine A dans des gouttelettes lipidiques. L’atteinte chronique du foie et la réaction inflammatoire qui y est associée provoquera la transition de ces cellules vers un phénotype myofibroblastique fibrogénique [4]. Les HSC activées produisent des protéines pro-inflammatoires, des médiateurs pro-fibrogènes et des inhibiteurs de dégradation de la matrice. Ainsi, l’apoptose ou l’altération des hépatocytes par le stress oxydant, le stress du RE et les altérations mitochondriales sont de puissants inducteurs de l’activation des HSC [4, 7, 8]. Enfin, les cellules de l’immunité innée (macrophages, cellules dendritiques), adaptative (lymphocytes B et lymphocytes T helper) et les cellules lymphoïdes innées, au cœur de l’inflammation hépatique, sont des acteurs incontournables du processus fibrogénique et de sa résolution [4, 7].
La cirrhose est le terrain privilégié du développement du carcinome hépatocellulaire (CHC). Néanmoins, la NASH constitue un facteur de risque en soi de développer un CHC, en dehors de toute cirrhose. L’inflammation, le stress oxydant et le stress du RE jouent un rôle majeur dans ce processus.
Dernière conséquence commune aux maladies chroniques du foie : le défaut de régénération hépatique. Le foie normal quiescent est en effet doué d’extraordinaires capacités régénératives qui se manifestent notamment lors d’une résection majeure d’un foie sain, la majorité des hépatocytes différenciés proliférant rapidement pour remplacer la masse hépatocytaire manquante [9]. En cas de stéatose, cette capacité proliférative des hépatocytes est drastiquement réduite. Par ailleurs, lors d’une agression chronique du foie, quel qu’en soit le vecteur, une réaction dite ductulaire (prolifération de cholangiocytes et/ou de cellules progénitrices), majoritairement composée de cellules biliaires, de cellules progénitrices hépatiques (HPC) et de cellules inflammatoires, apparaît autour de la veine porte [9]. Bien que les HPC aient la capacité de se différencier en hépatocytes et en cellules épithéliales biliaires, leur participation à la régénération chez l’homme reste probablement très limitée et leur implication dans la carcinogenèse hépatique a même été évoquée [10]. Comprendre les mécanismes responsables de ce défaut de régénération d’un foie stéatosique constitue donc un défi en pratique clinique [47] (→).
(→) Voir la Nouvelle de H. Gilgenkrantz, m/s n° 4, avril 2015, page 357
L’autophagie, arme d’autodestruction ou d’autodéfense ?
L’autophagie est un processus d’autodigestion permettant de maintenir l’homéostasie cellulaire par dégradation et recyclage de certains composants endommagés de la cellule (organites, protéines, métabolites, etc.). Il existe trois types d’autophagie canonique : la macro-autophagie, l’autophagie relayée par des protéines chaperonnes et la micro-autophagie.
La macro-autophagie est initiée par la formation d’une petite vésicule membranaire en forme de croissant appelée phagophore qui subit une étape d’élongation grâce à des protéines codées par les gènes de la famille ATG (autophagy-related genes) pour former un autophagosome. Ce dernier contient des composants cellulaires endommagés et des organites sénescents qui seront dégradés en fusionnant avec un lysosome. En bon citoyen assurant la valorisation des déchets, ce processus permettra ainsi aux acides aminés et aux AG libérés d’être recyclés ou réutilisés pour l’homéostasie énergétique de la cellule [48] (→).
(→) Voir la Nouvelle de P. Codogno, m/s 2004, n° 8-9, août-septembre 2004, page 734
Les mécanismes moléculaires impliqués dans les principales étapes de la macro-autophagie sont complexes et résumés dans la Figure 2 .
 |
Figure 2. Les trois types d’autophagie. 1. La macro-autophagie implique la formation d’une vésicule à double membrane appelée autophagosome. La formation du phagophore (étape d’initiation) est dépendante de kinases (ULK1, Unc-51-like autophagy activating kinase 1) et de protéines autophagiques dont ATG13 (autophagy-related gene 13). Un complexe incluant la Bécline-1 (ATG6) est impliqué dans l’étape de nucléation et d’élongation du phagophore. Le recrutement des protéines nécessaires à l’élongation et la fermeture de l’autophagosome impliquent ATG5-ATG12 et LC3 (ATG8)-PE (phosphatidyléthanolamine). La dernière phase implique la dégradation des protéines cargo, qui sont sélectivement reconnues par des adaptateurs comme p62, une protéine qui contient une région d’interaction avec LC3. 2. Dans l’autophagie relayée par les chaperonnes (CMA), les protéines qui contiennent un motif KFERQ se lient à la chaperonne Hsc70 et au récepteur LAMP-2A (lysosomal-associated membrane protein 2) pour être transloquées dans le lysosome et y être dégradées. 3. La microphagie implique une séquestration directe des composants cellulaires dans le lysosome par invagination. LC3 : microtubule-associated protein 1A/1B-light chain 3 ; p62 : sequestosome-1. |
La micro-autophagie séquestre directement certains constituants cellulaires dans le lysosome par invagination de sa membrane (Figure 2). Enfin, l’autophagie relayée par les chaperonnes (ou CMA) permet la dégradation de polypeptides contenant un motif KFERQ dans leur séquence. Ces substrats sont reconnus sélectivement par une protéine chaperonne de 70 kDa et transloqués dans le lysosome où ils sont dégradés par un complexe formé par la multimérisation de la protéine LAMP-2A (lysosome-associated membrane protein type 2A) [11] (Figure 2).
L’autophagie dans l’homéostasie métabolique du foie
Les fonctions métaboliques et de détoxification du foie font de cet organe un carrefour obligé du processus autophagique. La macro-autophagie et la CMA permettent, par exemple, d’éliminer les mitochondries endommagées (par mitophagie) [49] (→), d’éviter l’accumulation excessive d’agrégats protéiques intra-hépatocytaires et de recycler les nutriments dans un but d’économie et d’équilibre métaboliques. En conditions physiologiques, le jeûne est ainsi le principal inducteur d’autophagie. La restauration des concentrations sériques de glucose chez le nouveau-né [12], la gluconéogenèse, ou la formation des corps cétoniques, pendant les périodes de jeûne ainsi que la β-oxydation des AG, sont autant de fonctions physiologiques de l’autophagie hépatique. Les acides aminés utilisés pour la gluconéogenèse sont massivement produits par la protéolyse induite par l’autophagie, tandis que les AG sont issus d’une autophagie sélective des triglycérides stockés dans les gouttelettes lipidiques, encore appelée lipophagie [12]. Celle-ci permet également de contrôler le niveau des VLDL en libérant des AG et en dégradant l’apolipoprotéine B. En bref, l’autophagie est la gardienne d’un bon équilibre métabolique hépatique !
(→) Voir la Synthèse de P. Vigié et N. Camougrand, page 231 de ce numéro
L’autophagie hépatocytaire : un rempart contre la stéatose dans la NAFLD
Bien que les résultats des mesures de flux autophagique pratiquées in vivo chez l’animal et, a fortiori, dans les échantillons humains, soient à prendre avec précaution, il semble que le flux macro-autophagique soit diminué dans les foies stéatosiques [13]. L’étude d’une cohorte de patients obèses a en effet montré une augmentation du rapport LC3 (microtubule-associated protein 1A/1B-light chain 3)-II/LC3-I1 et des taux de p62/SQSTM1 (sequestosomes 2) intrahépatiques, témoin d’un blocage du flux autophagique, en cas de NAFLD et de NASH. Chez la souris, l’inhibition pharmacologique de la macro-autophagie de même que l’invalidation d’Atg5 ou d’Atg7 induisent un stress du RE et une stéatose hépatique [14, 15]. A contrario, une surexpression d’Atg7 réduit la stéatose hépatique, le stress du RE et l’insulinorésistance de souris soumises à un régime gras ou présentant une obésité génétiquement induite [15]. L’ensemble de ces résultats suggère que la macro-autophagie hépatocytaire protège contre la stéatose (Figure 3) [13].
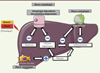 |
Figure 3. Le rôle de l’autophagie (CMA - autophagie relayée par les protéines chaperonnes - et macro-autophagie) dans les maladies chroniques du foie dépend du type cellulaire impliqué. La macro-autophagie (pour la NAFLD [non-alcoholic fatty liver disease] et l’ALD [maladie alcoolique du foie]) et la CMA (pour la NAFLD) dans l’hépatocyte protège de la stéatose et de l’apoptose en diminuant le stress cellulaire. Dans le macrophage, la macro-autophagie a des propriétés anti-inflammatoires et hépatoprotectrices, et son activation réduit par ce biais la fibrose. A contrario, la macro-autophagie a des propriétés pro-fibrogéniques dans les cellules étoilées. |
La CMA est également un régulateur essentiel du métabolisme des lipides et de la stéatose hépatique. En effet, les souris déficientes en récepteur LAMP-2A hépatocytaire développent une stéatose qui est encore majorée par un régime riche en graisses [16]. La baisse de l’oxydation mitochondriale des AG, de l’export des VLDL et une augmentation de la lipogenèse de novo participent à cette stéatose induite par le défaut de CMA (Figure 3).
La macro-autophagie dans l’ALD : avec le temps, va tout s’en va…
L’implication de l’autophagie dans la maladie alcoolique du foie, qu’elle soit aiguë ou chronique, est bien plus controversée. La consommation aiguë d’alcool semble augmenter la macro-autophagie dans les hépatocytes et diminuer l’apoptose et la stéatose hépatocytaires par dégradation des organites endommagés et des gouttelettes lipidiques [17]. Néanmoins, des résultats opposés, en particulier une augmentation des agrégats ubiquitinés associée à une diminution de la fonction des lysosomes et du flux macro-autophagique, ont été rapportés dans des modèles d’exposition chronique à l’alcool. Dans ce cas, l’inhibition de l’AMPK (5’ adenosine-monophosphate-activated protein kinase) par l’alcool pourrait diminuer la bêta-oxydation mitochondriale et augmenter la lipogenèse. La durée d’exposition jouerait ainsi un rôle essentiel.
Rôle anti-apoptotique et anti-inflammatoire de l’autophagie
Apoptose hépatocytaire et inflammation sont deux acteurs majeurs de la progression de la maladie, de la stéatose à la NASH. Dans l’hépatocyte, la macro-autophagie protège contre la mort cellulaire via l’élimination des protéines altérées, des lipides accumulés (par lipophagie) et des mitochondries endommagées (par mitophagie), permettant de diminuer le stress oxydant et de maintenir l’équilibre énergétique lors d’une agression hépatique [18]. La CMA protège également de l’atteinte hépatique, les souris déficientes en CMA présentant en effet des lésions majorées par un régime gras [16] (Figure 3).
Dans le macrophage, l’autophagie contrôle en particulier la phagocytose des pathogènes mais aussi leur maturation, de monocyte en macrophage [19]. Les macrophages dont le gène Atg5 a été inhibé sécrètent davantage d’IL(interleukine)-1β et d’IL-1α [20–22]. Chez la souris obèse, ils présentent un phénotype pro-inflammatoire, de type M1 [23]. Des lésions hépatiques, induites par le tétrachlorure de carbone, sont majorées chez les souris invalidées pour Atg5 spécifiquement dans la lignée myéloïde. Elles sont également associées à un recrutement accru de neutrophiles et de monocytes [24]. Bien que l’inhibition de la macro-autophagie dans les cellules myéloïdes n’ait aucun impact sur la maladie chronique du foie induite par l’alcool, son induction limite l’inflammation et la stéatose. Dans un modèle murin, le récepteur 2 des cannabinoïdes protège en effet des lésions induites par l’alcool en induisant un processus d’autophagie [25]. Tout semble donc indiquer que, dans la lignée macrophagique, la macro-autophagie a un effet anti-inflammatoire. Elle pourrait, par conséquent, protéger le foie d’une évolution vers la stéatohépatite.
Macro-autophagie et fibrose hépatique : tout dépend où se joue la pièce !
L’apoptose hépatocytaire, en activant les cellules étoilées du foie, est un élément initiateur de la fibrose hépatique. En protégeant du dommage cellulaire, la macro-autophagie hépatocytaire pourrait donc diminuer ce processus [26, 27]. Ce mécanisme pourrait être généralisé aux cellules épithéliales puisqu’il a également été retrouvé dans un autre contexte, celui de la fibrose rénale [28]. L’effet anti-fibrosant de l’autophagie macrophagique fait également intervenir son rôle anti-inflammatoire, deuxième combustible de la fibrogenèse. En effet, les souris déficientes en Atg5 spécifiquement dans la lignée myéloïde, présentent une fibrose exacerbée [24] et les myofibroblastes exposés à un milieu conditionné de macrophages dépourvus d’Atg5 ont un potentiel fibrogénique accru via la production augmentée d’IL-1α et d’IL-1β [24].
Les conséquences sont totalement différentes lorsque la cible de l’autophagie est la cellule étoilée dont l’activation vers un phénotype fibrogénique est marquée par la perte progressive de gouttelettes lipidiques enrichies en vitamine A. La lipophagie accrue associée à l’activation des HSC permet de générer des AG libres, d’augmenter la production d’ATP et de fournir l’énergie requise à la modification phénotypique des cellules fibrogéniques du foie. Les HSC humaines, ou murines, activées in vitro ou in vivo par une lésion hépatique présentent en effet une augmentation du rapport LC3-II/LC3-I, une baisse d’expression de p62 et un accroissement du flux autophagique [29, 30]. A contrario, l’inhibition de l’autophagie (qu’elle soit pharmacologique ou induite par des siARN [small interfering RNA] dirigés contre Atg5 ou Atg7) diminue le nombre de gouttelettes lipidiques dans ces cellules, leur prolifération et l’expression de gènes fibrogènes. De même, le foie des souris invalidées pour Atg7, spécifiquement dans les HSC, présente une diminution de la fibrose hépatique [29]. L’autophagie dans les cellules étoilées contribue donc à la fibrose hépatique en augmentant leurs propriétés pro-fibrogéniques. Ce mécanisme pourrait être généralisé à d’autres organes comme le rein, le cœur ou le poumon : l’inactivation de l’autophagie in vitro dans les cellules impliquées dans la fibrose rénale, pulmonaire ou cardiaque diminue en effet les propriétés fibrogéniques de ces cellules [29, 31] (Figure 3).
L’autophagie : un acteur essentiel de la régénération hépatique
L’autophagie étant impliquée dans la réorganisation intracellulaire des protéines et le maintien de l’homéostasie énergétique, il était logique de penser qu’elle puisse jouer un rôle important dans le processus de régénération hépatique. Le fait que la macro-autophagie soit induite dans les premières phases de la régénération hépatique après hépatectomie partielle (HP) renforce encore cette hypothèse [32]. Pourtant, très peu d’études expérimentales ont abordé cette question jusqu’à présent. L’invalidation hépato-spécifique d’Atg7 ou d’Atg5, chez la souris, entraîne des altérations mitochondriales, une apoptose hépatocytaire et un défaut de régénération du foie après HP [32–34]. Ainsi, l’autophagie protégerait les hépatocytes au cours de la régénération en permettant le maintien d’une production énergétique nécessaire et suffisante et en prévenant la sénescence hépatocytaire. De même, l’inhibition d’Atg5 dans les cellules progénitrices hépatiques (HPC) de rat inhibe leur différenciation hépatocytaire et leur capacité à participer à la régénération du foie lorsque celle des hépatocytes a préalablement été bloquée [35]. Cependant, les hépatocytes ne sont pas les seules cellules impliquées dans la régénération et aucune étude n’a porté jusqu’à présent sur le rôle de l’autophagie dans les cellules non parenchymateuses, ni sur son implication dans le défaut de régénération du foie stéatosique.
Autophagie et cancer du foie : des liaisons potentiellement dangereuses
Le rôle de l’autophagie dans le maintien de l’homéostasie cellulaire suggère qu’elle pourrait protéger les cellules d’une transformation maligne. En effet, de nombreuses caractéristiques de l’autophagie comme la limitation des ROS par mitophagie, le contrôle énergétique et métabolique - qui limite les besoins accrus des cellules cancéreuses -, l’immuno-surveillance ou la réponse anti-inflammatoire sont autant de processus suppresseurs de tumeur. Pourtant, certains travaux indiquent a contrario que l’autophagie pourrait favoriser la progression et l’invasion tumorales [36]. Qu’en est-il dans le foie ?
Différents arguments suggèrent que l’autophagie aurait un rôle protecteur vis-à-vis du carcinome hépatocellulaire (CHC). Chez l’homme, la diminution de l’activité autophagique dans les CHC, par rapport au tissu non tumoral, est corrélée à un mauvais pronostic [37]. Chez la souris, une délétion hétérozygote du gène codant la Bécline-1 (une des protéines de la famille des ATG) induit des cancers spontanés dans différents organes dont le foie [38]. De même, l’inhibition de la macro-autophagie, ou de la CMA, promeut le développement tumoral [39–41]. Cependant, dans ce cas, il s’agit d’adénomes bénins, ce qui suppose que d’autres événements génétiques soient requis pour induire la carcinogenèse. Les mécanismes de tumorigenèse induite par le défaut d’Atg7 impliquent l’accumulation de la protéine adaptatrice p62, dont la fonction principale est de conduire les protéines poly-ubiquitinées vers l’autophagosome pour leur dégradation. L’accumulation de p62 induit, entre autres, la stabilisation de la protéine NRF2 (nuclear factor erythroid related factor 2) en se liant à son inhibiteur Keap1 (Kelch-like ECH-associated protein 1). NRF2 module l’oxydo-réduction cellulaire et le métabolisme du glucose et de la glutamine favorisant ainsi la prolifération des cellules cancéreuses [42]. Bien que la contribution de l’inhibition de la macro-autophagie dans cette accumulation de p62 reste à être prouvée, des travaux récents du groupe de M. Karin ont démontré que p62 est nécessaire et suffisant pour l’induction de CHC chez la souris en impliquant le facteur NRF2 et la voie mTORC1 (mammalian target of rapamycin 1) [43].
Cependant, l’autophagie pourrait également promouvoir l’invasion tumorale [44]. En effet, l’inhibition des gènes codant la Bécline-1 ou Atg5 réduit le pouvoir métastatique pulmonaire de lignées de CHC chez la souris [45]. De plus, une induction du flux autophagique est décrite dans les CHC avancés en corrélation avec un mauvais pronostic.
L’autophagie pourrait donc jouer un double jeu : elle restreindrait le développement tumoral au départ, mais une fois celui-ci initié, elle permettrait aux cellules cancéreuses de survivre dans un microenvironnement qui leur est défavorable, devenant ainsi un acteur de la progression tumorale (Figure 4). Une approche induisant l’autophagie pourrait-elle avoir une place dans l’arsenal thérapeutique très restreint du CHC ? Les données expérimentales ayant abordé cette question sont contradictoires : certes, la rapamycine, un activateur majeur de l’autophagie, diminue la récurrence du CHC après transplantation [46] ; a contrario, l’inhibition de la CMA dans les cellules cancéreuses diminue leur tumorigénicité et leurs caractéristiques invasives.
 |
Figure 4. L’autophagie dans la carcinogenèse hépatique : un rôle protecteur dans l’initiation mais facilitateur dans l’invasion tumorale. ? L’inhibition de l’autophagie favorise la production de ROS (espèces réactives de l’oxygène) dues à la baisse de la mitophagie, et génère un stress génotoxique inducteur de mutations initiatrices de cancer. Cette inhibition de l’autophagie pourrait également conduire à l’accumulation de la protéine adaptatrice p62 qui induit l’hyper-activation des voies NRF2 (nuclear factor erythroid-related factor 2) et mTOR (mammalian target of rapamycin) et à l’initiation du processus tumoral. Une fois le processus oncogénique initié, l’activation de l’autophagie favoriserait au contraire l’invasion tumorale et la progression métastatique. ATG : autophagy-related genes ; Keap1 : Kelch-like ECH-associated protein 1. |
Conclusion
L’autophagie est essentielle au maintien de l’homéostasie hépatique et à la régénération du foie. Elle protège les hépatocytes contre l’accumulation de lipides intracellulaires et ses effets anti-inflammatoires et anti-apoptotiques au cours de l’évolution de la NAFLD et de l’ALD lui confèrent un caractère anti-fibrosant dès lors que le macrophage et l’hépatocyte constituent son terrain de jeu. Cependant, son action pro-fibrosante par activation des cellules étoilées du foie et son impact sur l’invasion et la progression tumorale du CHC nécessitent de rester vigilant sur son utilisation comme nouvelle cible thérapeutique.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Ces travaux cités ont été soutenus financièrement par l’Inserm, l’Université de Nice (PG), l’Université Paris-Diderot, le Labex Inflamex (SL), l’AFEF (PG, SL, HG), la Société Francophone du Diabète (PG), SFD/Roche Pharma (PG), la Fondation pour la Recherche Médicale (FRM DEQ20150331726 (SL)) et l’Agence nationale de la recherche (ANR-15-CE14-0016-01 [PG & SL], ANR-15-CE14-0031 [SL]).
La forme cytosolique de LC3 (LC3-I) se conjugue à la phosphatidyl-éthanolamine pour former LC3-II recrutée à la membrane de l’autophagosome.
Un récepteur des protéines ubiquitinées.
Références
- Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA 2015 ; 313 : 2263–2273. [CrossRef] [PubMed] [Google Scholar]
- Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol 2015 ; 12 : 231–242. [CrossRef] [PubMed] [Google Scholar]
- Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 2010 ; 52 : 774–788. [CrossRef] [PubMed] [Google Scholar]
- Mallat A, Lotersztajn S. Cellular mechanisms of tissue fibrosis. Novel insights into liver fibrosis. Am J Physiol Cell Physiol 2013 ; 305 : C789–C799. [CrossRef] [PubMed] [Google Scholar]
- Tran A, Gual P. Non-alcoholic steatohepatitis in morbidly obese patients. Clin Res Hepatol Gastroenterol 2013 ; 37 : 17–29. [Google Scholar]
- Marra F, Lotersztajn S. Pathophysiology of NASH: perspectives for a targeted treatment. Curr Pharm Des 2013 ; 19 : 5250–5269. [CrossRef] [PubMed] [Google Scholar]
- Lotersztajn S, Julien B, Texeira-Clerc F, et al. Hepatic fibrosis: molecular mechanisms and drug targets. Annu Rev Pharmacol Toxicol 2005 ; 45 : 605–628. [Google Scholar]
- Jiang JX, Mikami K, Venugopal S, et al. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-kappaB-dependent pathways. J Hepatol 2009 ; 51 : 139–148. [CrossRef] [PubMed] [Google Scholar]
- Gilgenkrantz H, Collin de l’Hortet A. New insights into liver regeneration. Clin Res Hepatol Gastroenterol 2011 ; 35 : 623–629. [Google Scholar]
- Dubuquoy L, Louvet A, Lassailly G, et al. Progenitor cell expansion and impaired hepatocyte regeneration in explanted livers from alcoholic hepatitis. Gut 2015 ; 64 : 1949–1960. [CrossRef] [PubMed] [Google Scholar]
- Arias E, CuervoA M. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol 2011 ; 23 : 184–189. [CrossRef] [PubMed] [Google Scholar]
- Madrigal-Matute J, Cuervo AM. Regulation of liver metabolism by autophagy. Gastroenterology 2016 ; 150 : 328–339. [CrossRef] [PubMed] [Google Scholar]
- Lavallard VJ, Gual P. Autophagy and non-alcoholic fatty liver disease. Biomed Res Int 2014 ; 2014 : 120179. [Google Scholar]
- Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature 2009 ; 458 : 1131–1135. [CrossRef] [PubMed] [Google Scholar]
- Yang L, Li P, Fu S, et al. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 2010 ; 11 : 467–478. [CrossRef] [PubMed] [Google Scholar]
- Schneider JL, Suh Y, Cuervo AM. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 2014 ; 20 : 417–432. [CrossRef] [PubMed] [Google Scholar]
- Ding WX, Manley S, Ni HM. The emerging role of autophagy in alcoholic liver disease. Exp Biol Med 2011 ; 236 : 546–556. [CrossRef] [PubMed] [Google Scholar]
- Czaja MJ. Function of autophagy in nonalcoholic fatty liver disease. Dig Dis Sci 2016 ; 61 : 1304–1313. [CrossRef] [PubMed] [Google Scholar]
- Jacquel A, Obba S, Boyer L, et al. Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions. Blood 2012 ; 119 : 4527–4531. [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011 ; 469 : 221–225. [CrossRef] [PubMed] [Google Scholar]
- Chuang SY, Yang CH, Chou CC, et al. TLR-induced PAI-2 expression suppresses IL-1β processing via increasing autophagy and NLRP3 degradation. Proc Natl Acad Sci USA 2013 ; 110 : 16079–16084. [CrossRef] [Google Scholar]
- Liao X, Sluimer JC, Wang Y, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 2012 ; 15 : 545–553. [CrossRef] [PubMed] [Google Scholar]
- Liu K, Zhao E, Ilyas G, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015 ; 11 : 271–284. [CrossRef] [PubMed] [Google Scholar]
- Lodder J, Denaës T, Chobert MN, et al. Macrophage autophagy protects against liver fibrosis in mice. Autophagy 2015 ; 11 : 1280–1292. [CrossRef] [PubMed] [Google Scholar]
- Denaës T, Lodder J, Chobert MN, et al. The cannabinoid receptor 2 protects against alcoholic liver disease via a macrophage autophagy-dependent pathway. Sci Rep 2016 ; 6 : 28806. [CrossRef] [PubMed] [Google Scholar]
- Mallat A, Lodder J, Teixeira-Clerc F, et al. Autophagy: a multifaceted partner in liver fibrosis. Biomed Res Int 2014 ; 2014 : 869390. [Google Scholar]
- Hidvegi T, Ewing M, Hale P, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 2010 ; 329 : 229–232. [Google Scholar]
- Li H, Peng X, Wang Y, et al. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy 2016 ; 12 : 1472–1486. [CrossRef] [PubMed] [Google Scholar]
- Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012 ; 142 : 938–946. [CrossRef] [PubMed] [Google Scholar]
- Thoen LF, Guimarães EL, Dollé L, et al. A role for autophagy during hepatic stellate cell activation. J Hepatol 2011 ; 55 : 1353–1360. [CrossRef] [PubMed] [Google Scholar]
- Ghavami S, Cunnington RH, Gupta S, et al. Autophagy is a regulator of TGF-β1-induced fibrogenesis in primary human atrial myofibroblasts. Cell Death Dis 2015 ; 6 : e1696. [CrossRef] [PubMed] [Google Scholar]
- Lin CW, Chen YS, Lin CC, et al. Amiodarone as an autophagy promoter reduces liver injury and enhances liver regeneration and survival in mice after partial hepatectomy. Sci Rep 2015 ; 5 : 15807. [CrossRef] [PubMed] [Google Scholar]
- Ni HM, Woolbright BL, Williams J, et al. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepaticautophagy. J Hepatol 2014 ; 61 : 617–625. [CrossRef] [PubMed] [Google Scholar]
- Toshima T, Shirabe K, Fukuhara T, et al. Suppression of autophagy during liver regeneration impairs energy charge and hepatocyte senescence in mice. Hepatology 2014 ; 60 : 290–300. [CrossRef] [PubMed] [Google Scholar]
- Xue F, Hu L, Ge R, et al. Autophagy-deficiency in hepatic progenitor cells leads to the defects of stemness and enhances susceptibility to neoplastic transformation. Cancer Lett 2016 ; 371 : 38–47. [Google Scholar]
- Lazova R, Camp RL, Klump V, et al. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis and poor outcome. Clin Cancer Res 2012 ; 18 : 370–379. [CrossRef] [PubMed] [Google Scholar]
- Ding ZB, Shi YH, Zhou J, et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res 2008 ; 68 : 9167–9175. [Google Scholar]
- Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003 ; 112 : 1809–1820. [CrossRef] [PubMed] [Google Scholar]
- Takamura A, Komatsu M, Hara T, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev 2011 ; 25 : 795–800. [CrossRef] [PubMed] [Google Scholar]
- Inami Y, Waguri S, Sakamoto A, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol 2011 ; 193 : 275–284. [CrossRef] [PubMed] [Google Scholar]
- Kon M, Kiffin R, Koga H, et al. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med 2011 ; 3 : 109–117. [Google Scholar]
- Mitsuishi Y, Taguchi K, Kawatani Y, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012 ; 22 : 60–79. [Google Scholar]
- Umemura A, He F, Taniguchi K, et al. p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell 2016 ; 29 : 935–948. [CrossRef] [PubMed] [Google Scholar]
- Li J, Yang B, Zhou Q, Wu Y, et al. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis 2013 ; 34 : 1343–1351. [CrossRef] [PubMed] [Google Scholar]
- Peng YF, Shi YH, Ding ZB, et al. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 2013 ; 9 : 2056–2068. [CrossRef] [PubMed] [Google Scholar]
- Toso C, Merani S, Bigam DL, et al. Sirolimus-based immuno-suppression is associated with increased survival after liver transplantation for hepatocellular carcinoma. Hepatology 2010 ; 51 : 1237–1243. [CrossRef] [PubMed] [Google Scholar]
- Gilgenkrantz H. Une seule cellule souche dans le foie : l’hépatocyte. Med Sci (Paris) 2015 ; 31 : 357–359. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Codogno P. Les gènes ATG et la macro-autophagie. Med Sci (Paris) 2004 ; 20 : 734–736. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Vigié P, Camougrand N. Mitophagie et contrôle qualité des mitochondries. Med Sci (Paris) 2017 ; 33 : 231–237. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Les maladies métaboliques chroniques du foie : ALD (alcoholic liver disease) et NAFLD (non-alcoholic liver disease) partagent des traits communs. L’obésité et la consommation chronique d’alcool sont responsables d’un spectre de lésions histologiques regroupées sous le terme de « stéatopathies », allant de l’accumulation de gras dans le foie (stéatose) à l’association de la stéatose et de l’inflammation (stéatohépatite) et à l’activation du processus fibrogénique conduisant à la cirrhose et à l’insuffisance hépatique associée à une hypertension portale et une encéphalopathie hépatique. La progression vers ces différentes étapes sont dépendantes non seulement de mécanismes intra- mais également extra-hépatiques, touchant notamment (1) l’intestin avec une dysbiose et une augmentation de la perméabilité intestinale participant à l’inflammation hépatique via les motifs moléculaires associés aux pathogènes (PAMP), dont le lipopolysaccharide (LPS), et (2) le tissu adipeux et son flux accru d’acides gras libres (AGL) et d’adipokines qui participent à l’apoptose hépatocytaire et à l’inflammation hépatique. Ces deux éléments sont essentiels au processus fibrogénique conduisant à la cirrhose. Chacune de ces étapes est associée à un défaut de régénération du foie. |
| Dans le texte | |
|
Figure 2. Les trois types d’autophagie. 1. La macro-autophagie implique la formation d’une vésicule à double membrane appelée autophagosome. La formation du phagophore (étape d’initiation) est dépendante de kinases (ULK1, Unc-51-like autophagy activating kinase 1) et de protéines autophagiques dont ATG13 (autophagy-related gene 13). Un complexe incluant la Bécline-1 (ATG6) est impliqué dans l’étape de nucléation et d’élongation du phagophore. Le recrutement des protéines nécessaires à l’élongation et la fermeture de l’autophagosome impliquent ATG5-ATG12 et LC3 (ATG8)-PE (phosphatidyléthanolamine). La dernière phase implique la dégradation des protéines cargo, qui sont sélectivement reconnues par des adaptateurs comme p62, une protéine qui contient une région d’interaction avec LC3. 2. Dans l’autophagie relayée par les chaperonnes (CMA), les protéines qui contiennent un motif KFERQ se lient à la chaperonne Hsc70 et au récepteur LAMP-2A (lysosomal-associated membrane protein 2) pour être transloquées dans le lysosome et y être dégradées. 3. La microphagie implique une séquestration directe des composants cellulaires dans le lysosome par invagination. LC3 : microtubule-associated protein 1A/1B-light chain 3 ; p62 : sequestosome-1. |
| Dans le texte | |
|
Figure 3. Le rôle de l’autophagie (CMA - autophagie relayée par les protéines chaperonnes - et macro-autophagie) dans les maladies chroniques du foie dépend du type cellulaire impliqué. La macro-autophagie (pour la NAFLD [non-alcoholic fatty liver disease] et l’ALD [maladie alcoolique du foie]) et la CMA (pour la NAFLD) dans l’hépatocyte protège de la stéatose et de l’apoptose en diminuant le stress cellulaire. Dans le macrophage, la macro-autophagie a des propriétés anti-inflammatoires et hépatoprotectrices, et son activation réduit par ce biais la fibrose. A contrario, la macro-autophagie a des propriétés pro-fibrogéniques dans les cellules étoilées. |
| Dans le texte | |
|
Figure 4. L’autophagie dans la carcinogenèse hépatique : un rôle protecteur dans l’initiation mais facilitateur dans l’invasion tumorale. ? L’inhibition de l’autophagie favorise la production de ROS (espèces réactives de l’oxygène) dues à la baisse de la mitophagie, et génère un stress génotoxique inducteur de mutations initiatrices de cancer. Cette inhibition de l’autophagie pourrait également conduire à l’accumulation de la protéine adaptatrice p62 qui induit l’hyper-activation des voies NRF2 (nuclear factor erythroid-related factor 2) et mTOR (mammalian target of rapamycin) et à l’initiation du processus tumoral. Une fois le processus oncogénique initié, l’activation de l’autophagie favoriserait au contraire l’invasion tumorale et la progression métastatique. ATG : autophagy-related genes ; Keap1 : Kelch-like ECH-associated protein 1. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.