")
")
| Issue |
Med Sci (Paris)
Volume 24, Number 2, Février 2008
|
|
|---|---|---|
| Page(s) | 185 - 190 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2008242185 | |
| Published online | 15 February 2008 | |
lschémie cérébrale
Modulations des récepteurs NMDA et mort neuronale retardée
Transient brain ischemia: NMDA receptor modulation and delayed neuronal death
1
UMR 6026-CNRS, Université de Rennes 1, France
2
Novartis Institutes for Biomedical Research, Basel, Suisse
3
Brain Research Institute, University of Zurich, Suisse
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
L’ischémie cérébrale transitoire induit une mort neuronale retardée de certains types cellulaires et régions cérébrales mais épargne des neurones d’autres territoires. Un processus clé par lequel la privation d’oxygène et de glucose déclenche la mort cellulaire est l’accumulation excessive de glutamate menant à une hyperexcitabilité neuronale. Cet article propose une mise à jour des connaissances sur les mécanismes neurophysiologiques cellulaires et moléculaires générant la plasticité post-ischémique des récepteurs NMDA. Nous la focaliserons sur l’étude des neurones pyramidaux CA1 vulnérables comparés aux neurones résistants de la région voisine CA3 au sein de l’hippocampe. Les modulations spécifiques des récepteurs NMDA pourraient expliquer la vulnérabilité sélective de certains types neuronaux. Les implications de ces mécanismes dans le traitement des attaques cérébrales et les raisons de l’échec de l’utilisation des antagonistes des récepteurs NMDA dans les essais cliniques humains sont également discutées.
Abstract
Transient global ischemia induces delayed neuronal death in certain cell types and brain regions while sparing cells in other areas. A key process through which oxygen-glucose deprivation triggers cell death is the excessive accumulation of the neurotransmitter glutamate leading to over excitation of neurons. In certain neurons this increase in glutamate will potentiate the NMDA type of glutamate receptor, which can then initiate cell death. This review provides an update of the neurophysiological, cellular and molecular mechanisms inducing postischemic plasticity of NMDA receptors, focusing on the sensitive CA1 pyramidal neurons in the hippocampus as compared to the relatively resistant neighboring CA3 neurons. Both a change in the equilibrium between protein tyrosine kinases/phosphatases and an increased density of surface NMDA receptors in response to ischemia may explain the selective vulnerability of specific cell types. Implications for the treatment of stroke and reasons for the failures of human clinical trials utilizing NMDA receptor antagonists are also discussed.
© 2008 médecine/sciences - Inserm / SRMS
L’ischémie cérébrale est la résultante d’une insuffisance ou d’un arrêt d’apport sanguin cérébral entraînant une suppression des apports en glucose et en oxygène. Elle peut être provoquée par un accident vasculaire cérébral (thrombose, compression ou rupture artérielle) et affecter ainsi un territoire délimité (ischémie focale) ou bien toucher tout le cerveau dans le cas d’un arrêt cardiaque temporaire (ischémie globale). La durée de l’ischémie et l’étendue des zones détériorées déterminent la gravité des répercussions cérébrales. Après un tel épisode, les lésions cérébrales continuent de s’étendre au cours du temps [1], et l’un des objectifs d’une prise en charge médicale consiste à essayer de limiter cette expansion.
Un épisode hypoxique-aglycémique provoque un dysfonctionnement neuronal quasi-immédiat puis une mort cellulaire consécutive très rapide si l’arrêt de perfusion dépasse la dizaine de minutes. Dans cette synthèse nous présentons les mécanismes cellulaires à l’origine de la dégénérescence neuronale retardée, et impliqués dans l’expansion de la lésion cérébrale. Ils ne concernent donc que les tissus touchés par une ischémie globale transitoire de durée limitée et en partie les tissus des zones péri-lésionnelles (zone dénommée pénombre) affectées à la suite d’une ischémie focale.
Vulnérabilité neuronale et mort retardée des neurones
Lors d’une ischémie globale temporaire (provoquée par exemple par un arrêt cardiaque transitoire de quelques minutes) deux phénomènes inattendus apparaissent (Figure 1):
- (1)
Toutes les zones du cerveau ne présentent pas la même vulnérabilité. La région préoptique, le cortex, l’hippocampe et le striatum sont des régions très sensibles à l’ischémie. Au sein d’une même structure cérébrale comme l’hippocampe, les neurones pyramidaux des régions CA1 meurent alors que les neurones pyramidaux de la région voisine CA3 survivent à une ischémie transitoire de quelques minutes. Il existe donc une vulnérabilité particulière de certaines structures cérébrales et plus particulièrement de certains types neuronaux [2, 3]. Réciproquement, cette observation indique que certains types de neurones ont développé des mécanismes neuroprotecteurs endogènes ou bien ont une constitution qui les rend moins sensibles. En comparant soigneusement la réponse ischémique distincte dans ces deux types cellulaires, il devrait donc être possible d’identifier les composants moléculaires qui sous-tendent cette protection endogène et de traduire ces résultats en applications cliniques.
- (2)
La mort de ces neurones n’est pas immédiate. En l’absence d’hémorragie, les lésions cérébrales de patients ayant subi une ischémie générale transitoire n’apparaissent qu’après trois à six jours sur des images de scanner X et un à deux jours en imagerie par résonance magnétique [4–6]. In vitro, après une hypoxie-aglycémie de quatre à six minutes, la mort des neurones vulnérables est constatée entre 24 et 72 h [7]. Cet intervalle constitue une fenêtre thérapeutique pendant laquelle des médicaments neuroprotecteurs pourraient, espérons-le, être capables de préserver les neurones de la mort cellulaire.
 |
Figure 1. Libération de glutamate pendant la phase ischémique et vulnérabilité cellulaire. Dans l’hippocampe de rat (photo du haut) les neurones pyramidaux, marqués ici à la biocytine, ne présentent pas la même sensibilité à l’ischémie transitoire. Pendant l’épisode ischémique, une réversion des transporteurs au glutamate de cellules gliales (en jaune) et neuronales (en rouge), engendre une forte élévation de glutamate extracellulaire (ronds verts). Le neuromédiateur va activer les récepteurs glutamatergiques post-synaptiques, notamment les récepteurs NMDA perméants au calcium à la fois dans la région CA3 et CA1 de l’hippocampe. Lorsque l’ischémie s’arrête, les niveaux de glutamate et de calcium reviennent à leurs niveaux de bases. Or 24 à 72 h après, les neurones CA1 dégénèrent - mort dépendante des récepteurs NMDA et du calcium - et les neurones CA3 survivent. |
Les mécanismes cellulaires expliquant cette vulnérabilité neuronale et la mort retardée sont complexes et seulement partiellement connus. De nombreux modèles expérimentaux utilisant des animaux existent et consistent généralement à provoquer des occlusions vasculaires in vivo ; in vitro, l’on supprime temporairement le glucose et l’oxygène du milieu extracellulaire.
Durant un épisode anoxique-aglycémique, l’impact biologique est extrêmement complexe. Une multitude de facteurs biologiques varient comme le pH intracellulaire, les taux de lactate, d’acides gras libres, de polyamines ; l’on observe aussi un dysfonctionnement mitochondrial et une libération massive de neuromédiateurs.
Glutamate et récepteurs NMDA
Le glutamate est le neuromédiateur excitateur majeur du système nerveux central et il peut se fixer sur ses récepteurs canaux (ionotropiques) ou sur ses récepteurs couplés à des voies de signalisation intracellulaires (métabotropiques) [8]. Les récepteurs glutamatergiques ionotropiques sensibles au N-méthyl-D-aspartate (NMDAR) sont indispensables à la potentialisation à long terme des synapses (LTP), une forme de plasticité physiologique cruciale dans l’apprentissage et la mémoire [9,10].
Un consensus existe autour du rôle des récepteurs NMDA dans l’ischémie cérébrale. Le blocage de ces récepteurs par des antagonistes spécifiques ou la chélation du calcium intracellulaire dans des modèles animaux empêche la mort neuronale induite par l’ischémie. Cela suggère que l’influx calcique à travers les récepteurs NMDA constitue le signal déclencheur de la mort neuronale [11]. Malheureusement, comme cela sera expliqué plus loin, l’utilisation, pourtant logique, de bloqueurs des récepteurs NMDA ne fonctionne pas dans le traitement de patients atteints d’attaque cérébrale.
Le mécanisme déclenché par l’ischémie commence à être bien compris. L’oxygène étant nécessaire à la synthèse aérobie du « carburant » cellulaire, l’adénosine triphosphate (ATP), l’épisode hypoxique-aglycémique entraîne une chute des niveaux d’ATP intracellulaire provoquant une réversion des transporteurs au glutamate [12]. Le glutamate libéré alors par les neurones et les cellules gliales va inonder massivement les compartiments extracellulaires de différentes zones cérébrales et activer les récepteurs glutamatergiques synaptiques et extra-synaptiques. L’activation des récepteurs de type NMDA provoque au niveau des neurones un influx massif de sodium et surtout de calcium, ce dernier étant à l’origine d’environ 10 % du courant. A long terme, un excès de calcium est excitotoxique puisqu’il augmente les taux de radicaux libres, active des protéases et des endonucléases, déclenche des voies de signalisation apoptotique ou bien mène à la nécrose [13] (Figure 1).
Cependant, dans l’hippocampe, ce mécanisme n’explique pas la vulnérabilité spécifique de certains types cellulaires, ni le décalage temporel de la mort cellulaire pour plusieurs raisons : (1) l’augmentation de glutamate extracellulaire et les élévations de calcium intracellulaire ont bien lieu pendant une ischémie transitoire mais sont réversibles. Ces niveaux redeviennent normaux au début de la phase post-ischémique et pourtant les neurones pyramidaux CA1 vont mourir du fait d’une suractivation de récepteurs NMDA, 24h-72 h plus tard.
(2) L’augmentation de glutamate extracellulaire et de calcium intracellulaire a lieu à la fois dans les régions CA3 et CA1 avec des amplitudes et des cinétiques quasi-similaires, ce qui n’explique pas la fragilité des neurones CA1 [14,15].
Si, dans le modèle de l’hippocampe de rat in vitro, des antagonistes NMDA sont ajoutés deux heures après la fin de l’ischémie transitoire, alors les neurones survivent. L’influx direct de calcium à travers les récepteurs durant l’ischémie n’est pas suffisant pour déclencher la mort neuronale. En revanche une modification à long terme des propriétés des récepteurs NMDA et de l’influx calcique tardif associé pourrait expliquer ce phénomène.
Plasticité à long terme des récepteurs NMDA
Y. Ben-Ari et V. Crepel ont été les premiers à découvrir une plasticité à long terme des synapses glutamatergiques déclenchée par une ischémie transitoire [16–18]. Une potentialisation des synapses excitatrices, ayant pour origine une augmentation anormale des courants à travers les récepteurs NMDA, a été retrouvée dans de nombreuses régions cérébrales, et notamment les plus vulnérables à l’ischémie transitoire comme les neurones CA1 de l’hippocampe, le striatum, le cortex [3, 17, 19] (Figure 2). Ce phénomène est observé dans des modèles animaux d’ischémie globale mais également dans la zone de pénombre suite à une ischémie focale [20].
 |
Figure 2. Hypothèse de la plasticité des récepteurs NMDA post-ischémique dans CA1 et CA3. Mécanisme possible de déclenchement d’une potentialisation à long terme des courants NMDA synaptiques et extrasynaptiques par l’ischémie. Dans les neurones pyramidaux CA3, à gauche, faisant partie des régions plus résistantes, l’ischémie réprime les courants NMDA diminuant ainsi la perméabilité calcique suite à la libération de glutamate. Dans les neurones pyramidaux CA1, à droite, l’augmentation des réponses NMDA post-ischémique est vraisemblablement due à la fois aux changements des propriétés biophysiques des récepteurs (symbolisé par un –P pour phosphorylation) mais aussi à l’insertion de nouveaux récepteurs dans la membrane (symbolisé par un nombre plus important de récepteurs insérés dans la membrane des neurones CA1). Après ces transformations, le glutamate libéré génèrera un influx calcique plus important qui finira, sur le long terme, par être excitotoxique pour les cellules pyramidales CA1. |
L’augmentation des réponses NMDA, mesurée par de nombreux autres groupes de recherche, est associée à des remaniements fonctionnels et morphologiques. Au niveau moléculaire, les récepteurs NMDA sont des hétérotrétramères constitués de sous unités NR1 obligatoires et d’autres sous-unités NR2A, NR2B, NR2C, NR2D et NR3 [21]. Après une ischémie, le nombre de récepteurs NMDA extrasynaptiques NR2B insérés dans la membrane augmente [20] et les réponses synaptiques NMDA s’amplifient parallèlement à la mise en place de sous unités NR1, dépendante de la synthèse protéique [22]. Des modifications structurales des neurones du réseau synaptique et des épines dendritiques [23, 24], consécutives à l’activation des NMDAR, sont constatées. Certains de ces changements morphologiques sont toutefois réversibles [25]. Chez le rat, un arrêt cardiaque de huit minutes diminue le nombre d’épines dendritiques dans CA1 et perturbe sélectivement la mémoire spatiale [26]. Dans CA1, une plasticité pathologique créée par l’ischémie transitoire englobe une augmentation des courants globaux NMDA, du nombre de leurs récepteurs présents à la membrane ainsi qu’une modification de leurs propriétés biophysiques, et enfin une augmentation du nombre de synapses (Figure 2). Parallèlement, des récepteurs glutamatergiques de type AMPA subissent également des modifications qui les rendent perméables au calcium [27], changent le Zn++ intracellulaire [28] et participent également au phénomène d’excitotoxicité.
Origine de la potentialisation des récepteurs NMDA dans CA1
La libération de glutamate par l’ischémie va co-activer les différentes classes de récepteurs au glutamate comme les récepteurs mGluR, AMPA, kainate et NMDA. L’activation de récepteurs métabotropiques et de la protéine GTPasique associée Gq va stimuler CAKβ/PYK2 déclenchant ainsi l’activation de Src [29]. Les kinases de type Src (SKF pour Src kinase family) appartiennent à une famille de protéines intracellulaires à activité tyrosine kinase dont cinq membres sont exprimés dans le SNC de mammifères - Src, Fyn, Yes, Lck et Lyn [29]. Or Src est capable de phosphoryler les résidus tyrosine de la partie intracellulaire du récepteur NMDA et d’augmenter sa conductance [29]. Par exemple, l’activation des récepteurs métabotropiques mGluR de groupe I et NMDA provoque une potentialisation de ces derniers qui passe par la stimulation de SKF [30].
L’ischémie induit une augmentation de phosphorylation des résidus tyrosine des sous-unités NR2A et NR2B, associée au recrutement de Src, Fyn and CAKβ au niveau des densités postsynaptiques PSD et l’activation de Src et CAKβ. L’ischémie induit l’activation de Src en quelques minutes et la phosphorylation à long terme des récepteurs NMDA. La phosphorylation des NMDAR par Src génère une augmentation de la quantité d’ions calcium et sodium passant à travers le canal [29] (Figure 3). Nous avons montré que rendre inactif Src avant une ischémie transitoire empêche à la fois la potentialisation des récepteurs NMDA et la mort neuronale à long terme des neurones CA1 dans l’hippocampe [15]. Enfin, les inhibiteurs de SKF suppriment l’excitotoxicité dépendante de la suractivation des NMDAR in vitro [15, 31] mais diminuent également la surface de lésion cérébrale post-ischémique in vivo [32, 33]. Ces résultats suggèrent une implication forte des modulations SKF-NMDAR dans le mécanisme physiopathologique de la mort neuronale induite par l’ischémie [29]. Cependant, en conditions physiologiques, l’interaction SKF-NMDA est indispensable au phénomène de potentialisation à long terme associé à l’apprentissage et la mémoire [9].
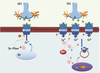 |
Figure 3. Schéma possible de plasticité post-ischémique des récepteurs NMDA. Des voies de signalisation distinctes seraient susceptibles de moduler différemment les récepteurs NMDA dans les neurones CA1 et CA3 de l’hippocampe. Dans CA3, à gauche, la montée de calcium intracellulaire (influx à partir du compartiment extracellulaire ou bien sortie de calcium à partir des stocks intracellulaires) pourraient déclencher un mécanisme protecteur visant à diminuer l’influx calcique à travers les récepteurs NMDA. Le calcium en tant que second messager peut activer des mécanismes de rétro-inhibition en activant indirectement des tyrosine phosphatases (Tyr-PTase) intracellulaires et d’autres voies inhibitrices calcium-dépendante qui vont diminuer les perméabilités calciques liées aux récepteurs NMDA. Ainsi, pendant la période de reperfusion, la libération de glutamate provoquera un influx calcique plus faible, évitant ainsi l’excitotoxicité. Dans CA1, la libération de glutamate pendant l’ischémie provoque une activation à long terme de Src, induisant la phosphorylation des résidus tyrosine du récepteur NMDA (symbolisé par un –P), ce qui augmente l’influx calcique à travers ces récepteurs. Cette modulation peut avoir lieu car la rétro-inhibition calcium-dépendante est potentiellement moins forte que dans CA3. De plus, il est fortement probable qu’une insertion de nouveaux récepteurs NMDA ait lieu dans la membrane plasmique au niveau synaptique et extrasynaptique à la suite de l’activation de voies de signalisation spécifiques. La plasticité à long terme physiopathologique induite par l’ischémie augmente ainsi la perméabilité calcique à travers les récepteurs NMDA et les risques d’excitotoxicité dans les neurones pyramidaux CA1. |
Pourquoi les neurones pyramidaux CA3 ne présentent-ils pas la même vulnérabilité que les neurones CA1 ?
Si la potentialisation synaptique post-ischémique est responsable de l’excitotoxicité, le changement de propriétés des récepteurs NMDA est-il spécifique aux neurones vulnérables ?
Pour répondre directement à cette question, notre groupe a décidé de mesurer l’impact d’une ischémie chimique transitoire (quatre minutes) sur des cultures organotypiques d’hippocampe de rat et d’en enregistrer les répercussions sur les propriétés des courants NMDA synaptiques et extrasynaptiques à la fois dans les cellules pyramidales CA1 et CA3 [15]. Les courants, enregistrés grâce à la méthode du patch-clamp, obtenus après une application extracellulaire localisée et répétitive de NMDA sont mesurés avant et après une brève période de privation en glucose et oxygène.
Une potentialisation post-ischémique des courants NMDA est observée dans les cellules pyramidales CA1 alors que les cellules CA3 « résistantes » présentent une dépression [15] (Figure 2). La corrélation entre vulnérabilité cellulaire et potentialisation des courants NMDA est donc maintenue.
Néanmoins, tous les éléments clés de la potentialisation existent également dans les neurones pyramidaux CA3 (récepteurs métabotropiques, Gq, interactions possibles SKF-NMDAR) et pourtant la potentialisation post-ischémique n’apparaît pas. Pendant l’ischémie, la libération de glutamate et la montée transitoire de calcium sont très similaires à celles qui sont observées en CA1, il est donc probable que le glutamate extracellulaire active les voies mGluR –Src –NMDA dans CA3. Or l’activation des voies de signalisation mGluR, PKC, Src génère une augmentation des courants NMDA de neurones CA3 [30].
La dépression post-ischémique enregistrée dans les neurones CA3 renforce encore l’idée de corrélation entre l’amplitude des courants NMDA et la vulnérabilité neuronale. La dépression des réponses globales peut être liée à une diminution du nombre de récepteurs présents à la membrane, à une modification de leurs propriétés biophysiques ou à l’association des deux. Donc, pendant une ischémie, un système d’inhibition très fort se met en place dans CA3.
La clé de l’énigme viendrait-elle d’une différence de modulation intracellulaire des récepteurs NMDA dans les 2 types cellulaires ? Nous avons comparé la sensibilité au calcium intracellulaire de ces récepteurs en enregistrant les courants et en utilisant simultanément l’imagerie calcique. L’élévation de calcium modifie peu les courants NMDA de CA1 mais déprime fortement les courants dans CA3 [34] (Figure 3). Il existe donc dans les neurones pyramidaux CA3 un système d’inhibition calcium-dépendant rétroactif, beaucoup plus efficace que dans CA1 [34].
En effet, si dans CA3 le système inhibiteur calcium-dépendant est bloqué, alors la dépression post-ischémique des récepteurs NMDA est transformée en potentialisation. Le même phénomène est constaté si les mGluR sont sélectivement activés : l’effet sera une augmentation ou au contraire une diminution des courants, en fonction des taux de tampons calciques utilisés. Comme pour la potentialisation à long terme, les niveaux de calcium intracellulaires vont déterminer le sens de la plasticité des réponses NMDA [35]. La rétroinhibition calcium-dépendante peut passer par l’activation d’une tyrosine phosphatase qui s’oppose à l’effet de Src [15, 36] (Figure 3).
Pourquoi un tel système, potentiellement protecteur, de rétro-inhibition n’est-il pas aussi présent dans CA1 ? Pour la LTP (long-term potentiation) physiologique, forme de plasticité cruciale dans l’apprentissage et la mémoire [9], l’élévation de calcium intracellulaire et l’influx calcique à travers les récepteurs NMDA sont absolument indispensables. Une rétro-inhibition calcium dépendante trop forte pourrait entraver ce mécanisme.
Conclusion et problématique thérapeutique
Les antagonistes NMDA fonctionnent bien dans les différents modèles animaux d’ischémie cérébrale. Ils empêchent la dégénérescence neuronale ou réduisent la zone péri-lésionnelle in vitro et in vivo. Cependant, la plupart des essais cliniques utilisant des antagonistes NMDA ont été abandonnés du fait de leur inefficacité ou de leurs effets secondaires majeurs.
Les différences entre les modèles animaux et la clinique humaine sont certainement multifactorielles. (1) En clinique, dans les nombreux cas d’ischémies focales, l’arrêt de perfusion est très souvent permanent suite, par exemple, à une rupture vasculaire. La zone de lésion présente alors une mort neuronale rapide, par nécrose, et les modifications des propriétés des glutamatergiques et de l’influx calcique n’y sont qu’un phénomène additionnel. (2) Chez l’homme, on ne peut agir qu’a posteriori. Les patients arrivent très tardivement à l’hôpital après l’AVC alors que dans les modèles animaux, les antagonistes sont appliqués rapidement après l’ischémie. Le délai d’application semble être critique [37]. (3) Bloquer les récepteurs NMDA revient à toucher l’ensemble des synapses glutamatergiques et donc à perturber également le fonctionnement des zones saines. Or, la plupart des fonctions cérébrales font intervenir des synapses glutamatergiques. Des effets secondaires cognitifs majeurs peuvent passer inaperçus dans les modèles animaux. (4) Si la mort neuronale est due à une plasticité pathologique qui crée des modifications morphologiques et des transformations à long terme du nombre, de l’activation et des propriétés des récepteurs NMDA, il est peu probable qu’un traitement de durée limitée puisse empêcher une mort neuronale. (5) Cibler les modulations aberrantes du NMDAR pour les ramener à leur état basal physiologique pourrait rétablir la situation mais les acteurs moléculaires de la régulation (mGluR de groupe I, PKC, NOS, MEK, ERK, CaKβ/PYK2) sont impliqués dans de nombreuses fonctions cérébrales, telles que l’apprentissage et la mémoire. En effet les voies de signalisation contrôlant la potentialisation ischémique et la potentialisation à long terme physiologique sont très proches. Une étude comparative plus approfondie de ces deux mécanismes de plasticité neuronale permettra peut-être à l’avenir d’empêcher sélectivement la mort cellulaire post-ischémique retardée.
Références
- Back T, Hemmen T, Schuler OG. Lesion evolution in cerebral ischemia. J Neurol 2004; 251 : 388–97. [Google Scholar]
- Pulsinelli WA. Selective neuronal vulnerability: morphological and molecular characteristics. Prog Brain Res 1985; 63 : 29–37. [Google Scholar]
- Calabresi P, Centonze D, Pisani A, et al. Synaptic plasticity in the ischaemic brain. Lancet Neurol 2003; 2 : 622–9. [Google Scholar]
- Rosenberg GA. Ischemic brain edema. Prog Cardiovasc Dis 1999; 42 : 209–16. [Google Scholar]
- Fujioka M, Taoka T, Hiramatsu KI, et al. Delayed ischemic hyperintensity on T1-weighted MRI in the caudoputamen and cerebral cortex of humans after spectacular shrinking deficit. Stroke 1999; 30 : 1038–42. [Google Scholar]
- Petito CK, Feldmann E, Pulsinelli WA, Plum F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology 1987; 37 : 1281–6. [Google Scholar]
- Kirino, T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 1982; 239 : 57–69. [Google Scholar]
- Galvez T, Pin JP. How do G-protein-coupled receptors work ? The case of metabotropic glutamate and GABA receptors. Med Sci (Paris) 2003; 19 : 559–65. [Google Scholar]
- MacDonald JF, Jackson MF, Beazely MA. Hippocampal long-term synaptic plasticity and signal amplification of NMDA receptors. Crit Rev Neurobiol 2006; 18 : 71–84. [Google Scholar]
- Spedding M, Lestage P. Synaptic plasticity and neuropathology: new approaches in drug discovery. Med Sci (Paris) 2005; 21 : 104–9. [Google Scholar]
- Collins RC, Dobkin BH, Choi DW. Selective vulnerability of the brain: new insights into the pathophysiology of stroke. Ann Intern Med 1989; 110 : 992–1000. [Google Scholar]
- Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 2000; 403 : 316–21. [Google Scholar]
- Choi DW. Excitotoxic cell death. J Neurobiol 1992; 23 : 1261–76. [Google Scholar]
- Mitani A, Andou Y, Kataoka K. Selective vulnerability of hippocampal CA1 neurons cannot be explained in terms of an increase in glutamate concentration during ischemia in the gerbil: brain microdialysis study. Neuroscience 1992; 48 : 307–13. [Google Scholar]
- Gee CE, Benquet P, Raineteau O, et al. NMDA receptors and the differential ischemic vulnerability of hippocampal neurons. Eur J Neurosci 2006; 23 : 2595–603. [Google Scholar]
- Crepel V, Hammond C, Chinestra P, et al. A selective LTP of NMDA receptor-mediated currents induced by anoxia in CA1 hippocampal neurons. J Neurophysiol 1993; 70 : 2045–55. [Google Scholar]
- Crepel V, Hammond C, Krnjevic K, et al. Anoxia-induced LTP of isolated NMDA receptor-mediated synaptic responses. J Neurophysiol 1993; 69 : 1774–8. [Google Scholar]
- Crepel V, Epsztein J, Ben-Ari Y. Ischemia induces short- and long-term remodeling of synaptic activity in the hippocampus. J Cell Mol Med 2003; 7 : 401–7. [Google Scholar]
- Calabresi P, Saulle E, Centonze D, et al. Post-ischaemic long-term synaptic potentiation in the striatum: a putative mechanism for cell type-specific vulnerability. Brain 2002; 125 : 844–60. [Google Scholar]
- Picconi B, Tortiglione A, Barone I, et al. NR2B subunit exerts a critical role in postischemic synaptic plasticity. Stroke 2006; 37 : 1895–901. [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 2001; 11 : 327–35. [Google Scholar]
- Quintana P, Alberi S, Hakkoum D, Muller D. Glutamate receptor changes associated with transient anoxia/hypoglycaemia in hippocampal slice cultures. Eur J Neurosci 2006; 23 : 975–83. [Google Scholar]
- Kovalenko T, Osadchenko I, Nikonenko A, et al. Ischemia-induced modifications in hippocampal CA1 stratum radiatum excitatory synapses. Hippocampus 2006; 16 : 814–25. [Google Scholar]
- Jourdain P, Nikonenko I, Alberi S, Muller D. Remodeling of hippocampal synaptic networks by a brief anoxia-hypoglycemia. J Neurosci 2002; 22 : 3108–16. [Google Scholar]
- Zhang S, Boyd J, Delaney K, Murphy TH. Rapid reversible changes in dendritic spine structure in vivo gated by the degree of ischemia. J Neurosci 2005; 25: 5333–8. [Google Scholar]
- Neigh GN, Glasper ER, Kofler J, et al. Cardiac arrest with cardiopulmonary resuscitation reduces dendritic spine density in CA1 pyramidal cells and selectively alters acquisition of spatial memory. Eur J Neurosci 2004; 20 : 1865–72. [Google Scholar]
- Tanaka H, Grooms SY, Bennett MV, Zukin RS. The AMPAR subunit GluR2: still front and center-stage. Brain Res 2000; 886 : 190–207. [Google Scholar]
- Noh KM, Yokota H, Mashiko T, et al. Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc Natl Acad Sci USA 2005; 102 : 12230–5. [Google Scholar]
- Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 2004; 5 : 317–28. [Google Scholar]
- Benquet P, Gee CE, Gerber U. Two distinct signaling pathways upregulate NMDA receptor responses via two distinct metabotropic glutamate receptor subtypes. J Neurosci 2002; 22 : 9679–86. [Google Scholar]
- Hashimoto R, Fujimaki K, Jeong MR, et al. Lithium-induced inhibition of Src tyrosine kinase in rat cerebral cortical neurons: a role in neuroprotection against N-methyl-D-aspartate receptor-mediated excitotoxicity. FEBS Lett 2003; 538 : 145–8. [Google Scholar]
- Ardizzone TD, Zhan X, Ander BP, Sharp FR. SRC kinase inhibition improves acute outcomes after experimental intracerebral hemorrhage. Stroke 2007; 38 : 1621–5. [Google Scholar]
- Paul R, Zhang ZG, Eliceiri BP, et al. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat Med 2001; 7 : 222–7. [Google Scholar]
- Grishin AA, Gee CE, Gerber U, Benquet P. Differential calcium-dependent modulation of NMDA currents in CA1 and CA3 hippocampal pyramidal cells. J Neurosci 2004; 24 : 350–5. [Google Scholar]
- Harney SC, Rowan M, Anwyl R. Long-term depression of NMDA receptor-mediated synaptic transmission is dependent on activation of metabotropic glutamate receptors and is altered to long-term potentiation by low intracellular calcium buffering. J Neurosci 2006; 26 : 1128–32. [Google Scholar]
- Grishin AA, Benquet P, Gerber U. Muscarinic receptor stimulation reduces NMDA responses in CA3 hippocampal pyramidal cells via Ca2+-dependent activation of tyrosine phosphatase. Neuropharmacology 2005; 49 : 328–37. [Google Scholar]
- Birmingham, K. Future of neuroprotective drugs in doubt. Nat Med 2002; 8 : 5. [Google Scholar]
Liste des figures
|
Figure 1. Libération de glutamate pendant la phase ischémique et vulnérabilité cellulaire. Dans l’hippocampe de rat (photo du haut) les neurones pyramidaux, marqués ici à la biocytine, ne présentent pas la même sensibilité à l’ischémie transitoire. Pendant l’épisode ischémique, une réversion des transporteurs au glutamate de cellules gliales (en jaune) et neuronales (en rouge), engendre une forte élévation de glutamate extracellulaire (ronds verts). Le neuromédiateur va activer les récepteurs glutamatergiques post-synaptiques, notamment les récepteurs NMDA perméants au calcium à la fois dans la région CA3 et CA1 de l’hippocampe. Lorsque l’ischémie s’arrête, les niveaux de glutamate et de calcium reviennent à leurs niveaux de bases. Or 24 à 72 h après, les neurones CA1 dégénèrent - mort dépendante des récepteurs NMDA et du calcium - et les neurones CA3 survivent. |
| Dans le texte | |
|
Figure 2. Hypothèse de la plasticité des récepteurs NMDA post-ischémique dans CA1 et CA3. Mécanisme possible de déclenchement d’une potentialisation à long terme des courants NMDA synaptiques et extrasynaptiques par l’ischémie. Dans les neurones pyramidaux CA3, à gauche, faisant partie des régions plus résistantes, l’ischémie réprime les courants NMDA diminuant ainsi la perméabilité calcique suite à la libération de glutamate. Dans les neurones pyramidaux CA1, à droite, l’augmentation des réponses NMDA post-ischémique est vraisemblablement due à la fois aux changements des propriétés biophysiques des récepteurs (symbolisé par un –P pour phosphorylation) mais aussi à l’insertion de nouveaux récepteurs dans la membrane (symbolisé par un nombre plus important de récepteurs insérés dans la membrane des neurones CA1). Après ces transformations, le glutamate libéré génèrera un influx calcique plus important qui finira, sur le long terme, par être excitotoxique pour les cellules pyramidales CA1. |
| Dans le texte | |
|
Figure 3. Schéma possible de plasticité post-ischémique des récepteurs NMDA. Des voies de signalisation distinctes seraient susceptibles de moduler différemment les récepteurs NMDA dans les neurones CA1 et CA3 de l’hippocampe. Dans CA3, à gauche, la montée de calcium intracellulaire (influx à partir du compartiment extracellulaire ou bien sortie de calcium à partir des stocks intracellulaires) pourraient déclencher un mécanisme protecteur visant à diminuer l’influx calcique à travers les récepteurs NMDA. Le calcium en tant que second messager peut activer des mécanismes de rétro-inhibition en activant indirectement des tyrosine phosphatases (Tyr-PTase) intracellulaires et d’autres voies inhibitrices calcium-dépendante qui vont diminuer les perméabilités calciques liées aux récepteurs NMDA. Ainsi, pendant la période de reperfusion, la libération de glutamate provoquera un influx calcique plus faible, évitant ainsi l’excitotoxicité. Dans CA1, la libération de glutamate pendant l’ischémie provoque une activation à long terme de Src, induisant la phosphorylation des résidus tyrosine du récepteur NMDA (symbolisé par un –P), ce qui augmente l’influx calcique à travers ces récepteurs. Cette modulation peut avoir lieu car la rétro-inhibition calcium-dépendante est potentiellement moins forte que dans CA3. De plus, il est fortement probable qu’une insertion de nouveaux récepteurs NMDA ait lieu dans la membrane plasmique au niveau synaptique et extrasynaptique à la suite de l’activation de voies de signalisation spécifiques. La plasticité à long terme physiopathologique induite par l’ischémie augmente ainsi la perméabilité calcique à travers les récepteurs NMDA et les risques d’excitotoxicité dans les neurones pyramidaux CA1. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.