")
")
| Issue |
Med Sci (Paris)
Volume 18, Number 1, Janvier 2002
|
|
|---|---|---|
| Page(s) | 86 - 90 | |
| Section | M/S Revues : Mini-Synthéses | |
| DOI | https://doi.org/10.1051/medsci/200218186 | |
| Published online | 15 January 2002 | |
Génotoxicité des métabolites des œstrogènes et cancers
Inserm U490, Toxicologie moléculaire, Faculté de médecine, 45, rue des Saints-Pères, 75270 Paris Cedex 06, France
L’exposition prolongée aux œstrogènes est un facteur qui favorise le développement de certains cancers, en particulier les cancers du sein. La stimulation de la croissance cellulaire par l’activation chronique des récepteurs des œstrogènes est le mécanisme le plus souvent évoqué. Depuis quelques années, certains métabolites génotoxiques de l’œstradiol, en particulier les catéchols et leurs dérivés quinoliques, sont suspectés comme agents de tumorigenèse. Ces différents métabolites n’ont pas la même toxicité et, tout facteur exogène, comme certains polluants, ou endogène comme les œstrogènes eux-mêmes, susceptible de modifier l’activité des enzymes les produisant, pourrait jouer un rôle dans la pathogénie de ces cancers.
© 2002 médecine/sciences - Inserm / SRMS
L’incidence du cancer du sein augmente chaque année dans les pays industrialisés. Il représente un tiers des cas de nouveaux cancers aux États-Unis chez les femmes, la première cause de mort par cancer chez les non fumeuses et la seconde chez les fumeuses. Si certaines mutations rares affectant des gènes, comme les suppresseurs de tumeurs BRCA1 et BRCA2, augmentent considérablement les risques de survenue de cancer du sein, l’un des problèmes attenant à cette maladie est d’isoler un agent étiologique commun à une grande partie des populations touchées. L’exposition prolongée aux œstrogènes constitue un facteur favorisant le développement des cancers mammaires qui sont, pour une grande part, hormono-dépendants. Deux mécanismes principaux ont été évoqués pour expliquer le rôle potentiel de ces molécules dans la carcinogenèse : le premier est la stimulation de la croissance des tumeurs par l’activation des récepteurs des œstrogènes. Le second implique les métabolites génotoxiques dérivés des œstrogènes, dont la synthèse dépend de l’expression et de l’activité de nombreuses enzymes.
Le premier mécanisme, considéré comme le principal par la plupart des auteurs, s’appuie sur des observations expérimentales qui ont été largement commentées. Cependant, de nombreuses données récentes semblent donner de plus en plus de crédit au rôle joué par les métabolites génotoxiques de l’œstradiol. Les dernières données sur le métabolisme de cette hormone seront revues dans cet article.
Métabolisme oxydatif des œstrogènes
Les œstrogènes dérivent du cholestérol et sont au nombre de trois, l’œstrone (E1), l’œstradiol (E2) qui est le principal œstrogène (Figure 1), et l’œstriol (E3). Leur squelette comporte 17 carbones et se caractérise par un cycle phénolique dont la position hydroxylée est le carbone 3 (C3). Différentes voies métaboliques peuvent conduire à la synthèse de composés génotoxiques. La voie principale (Figure 1) conduit à la synthèse de dérivés hydroxylés de l’E2, les catéchols. Il s’agit du 2CE (ou 2OH-E2 pour 20H- catechol estrogen) qui est le principal catéchol détecté à la fois dans le sang et dans l’urine, et du 4CE (ou 4OH-E2 pour 40H-catechol estrogen) produit de façon majoritaire dans certains tissus dont les tissus mammaire (tumoral ou non), ovarien, et l’endomètre. Ces composés sont synthétisés majoritairement par des cytochromes P450 (CYP) extrahépatiques ou hépatiques, mais aussi par d’autres enzymes comme l’aromatase ou certaines peroxydases [1,2]. Les différences de production des catéchols selon les tissus sont expliquées par le fait que l’expression des différents cytochromes P450 est variable selon les tissus. Ainsi, l’œstradiol est converti en 2CE par les CYP1A dans les tissus extrahépatiques et les CYP3A dans le foie. Ces enzymes ont aussi une activité 4-hydroxylase non spécifique permettant la synthèse en faible quantité (15 % environ) du 4CE [3]. Cependant, la voie principale de synthèse de cette autre forme de catéchol met en jeu le CYP1B1, exprimé dans la plupart des tissus à l’exception du foie [4,5].
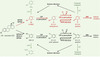 |
Figure 1. Voies métaboliques de l’œstradiol. L’œstradiol (E2, à gauche sur la figure) est le principal œstrogène. Il est métabolisé dans les cellules par différents types de cytochromes P450 (CYP) dont l’expression tissulaire est variable et, dans certains cas, inductible. Les produits de ces réactions sont les catéchols, qui, par action d’autres CYP ou de peroxydases, peuvent former des semi-quinones et des quinones. La formation des quinones peut être non enzymatique et aboutir à la formation d’ion superoxyde O2°-, dérivé réactif de l’oxygène très toxique. La présence de la P450 réductase permet un cycle entre semi-quinones et quinones qui, bien qu’elles soient présentes en petite quantité, conduit souvent à la formation de dérivés réactifs de l’oxygène. Les quinones sont les produits terminaux de la réaction et présentent une affinité pour l’ADN (toxique dans le cas de la E2-3,4-quinone). Différentes enzymes de détoxication (COMT, …) interviennent tout au long de la voie métabolique avec, pour rôle principal, d’inactiver l’une des fonctions hydroxyles des dérivés de l’œstradiol. Sont représentées respectivement en vert et en rouge les voies ou molécules dont le rôle protecteur ou toxique est avéré (inspirée de [24]). |
Dans un second temps, les catéchols, de par la position de leurs hydroxyles, peuvent conduire à la synthèse de semi-quinones puis, selon un mécanisme de réaction en chaîne, à celle des quinones (Figure 1). La formation des semi-quinones est un processus enzymatique impliquant des CYP ou des peroxydases. En revanche, celle des quinones peut se faire par plusieurs mécanismes, enzymatique (peroxydases ou CYP) ou non enzymatique impliquant dans ce cas l’oxygène moléculaire (O2) et conduisant à la formation d’ion superoxyde, O2°-.
Comme dans de nombreuses réactions, les différents composés toxiques dérivant de l’E2 sont eux-mêmes métabolisés par d’autres enzymes. La voie principale de détoxication des catéchols fait intervenir la COMT ou catéchol O-méthyl transférase qui méthyle les fonctions hydroxyles des catéchols pour former le 2MeCE et le 4MeCE. Cette méthylation bloque les fonctions hydroxyles et empêche la formation des semi-quinones et des quinones. D’autres enzymes de conjugaison, comme les sulfotransférases et les UDP-glucuronosyltransférases, contribuent de la même façon à ces processus de détoxication [6]. Quant aux quinones, elles peuvent être conjuguées au glutathion intracellulaire sous l’effet des glutathion-S-transférases (GST). Ces activités de conjugaison et de méthylation peuvent être contrebalancées par des activités inverses de déconjugaison (glucuronidases) et de déméthylation (déméthylases encore inconnues) [7].
Enfin, certaines enzymes comme la NADPH quinone réductase reforment des catéchols à partir des produits finaux de la réaction, les quinones [8].
Génotoxicité des dérivés oxydés des œstrogènes
Parmi les métabolites précédemment décrits, seuls les catéchols présentent une faible affinité pour le récepteur des œstrogènes. La liaison des quinones n’a pas été étudiée. Toutefois, compte tenu du fait que les concentrations de ces dérivés sont faibles, cette activité de liaison est probablement négligeable.
Les dérivés oxydés des œstrogènes stimulent malgré tout la croissance des tumeurs mammaires in vitro et in vivo (catéchols) et surtout, peuvent se lier à l’ADN et aux protéines (génotoxicité des quinones) [9]. La production des catéchols dépend des activités des enzymes qui les produisent, qui elles-mêmes dépendent du tissu étudié et de la présence ou non d’inducteurs. Lorsque la synthèse de catéchols devient excessive, les systèmes de détoxication (COMT, sulfotransférases, UDP-glucuronosyltransférases) sont dépassés et les dérivés, semiquinones et quinones, sont produits. La deuxième barrière de protection est la conjugaison des quinones grâce aux GST. Quand les réserves en glutathion s’épuisent, les quinones peuvent exercer leur génotoxicité. Elles sont en effet hautement réactives et capables d’une part de former des adduits d’ADN et des sites apuriniques, et d’autre part, d’oxyder les lipides cellulaires et certains ions métalliques (fer et cuivre) [10]. La formation des quinones à partir des semiquinones peut conduire à la formation de dérivés réactifs de l’oxygène comme l’ion superoxyde O2°-ainsi qu’à des cycles “futiles” d’oxydoré-duction (même en présence de faibles quantités de semi-quinones et de quinones) [11]. La production excessive de ces dérivés réactifs peut avoir des conséquences délétères pour es cellules car ces molécules endommagent l’ADN, les ipides et les protéines.
Un point important est la différence de toxicité des deux formes de catéchols. Ainsi, le 2CE serait moins toxique, voire en partie protecteur (le dérivé méthylé du 2CE inhibe la synthèse du 4CE), tandis que le 4CE aurait clairement un rôle génotoxique. En effet, seule l’injec-tion intrapéritonéale à des rongeurs de 4CE et de l’E2 a la capacité de provoquer le développement de tumeurs du rein [12, 13]. L’apparition de ces tumeurs a été attribuée à la formation d’adduits d’ADN (4OH-E2-1α,ß-N7-guanine et 4OH-E2-1(α,ß)-N3-adénosine) qui sont détectés aussi bien in vivo, qu’après traitement de lignées cellulaires in vitro [14]. D’autres arguments indirects suggèrent que le 4CE a un rôle génotoxique in vivo : ce métabolite n’est pas le catéchol majoritairement produit par l’organisme, mais est en revanche synthétisé par des tissus particuliers comme le sein, l’ovaire ou l’endomètre, où la forme 2CE est minoritaire [15, 16]. Or, ce sont ces tissus qui sont sensibles à la cancérogenèse induite par les œstrogènes. En outre, dans le tissu mammaire ou ovarien tumoral, une activité 4-hydroxylase plus élevée que dans le tissu normal est détectée [15, 16], et cette activité semble principalement due au CYP1B1, qui constitue la voie principale de synthèse du 4CE.
La faible toxicité du 2CE comparée à celle du 4CE peut s’expliquer par plusieurs mécanismes. Tout d’abord, les adduits d’ADN formés par le 2CE sont stables et peu mutagènes, contrairement à ceux provoqués par le 4CE [17]. Par ailleurs, le 2CE est plus rapidement inactivé par méthylation que le 4CE en raison d’une affinité plus importante de la COMT pour ce métabolite [18]. Enfin, la forme méthylée dérivée du 2CE, produite par la COMT, pourrait inhiber la production de 4CE, ce qui renforce encore son caractère protecteur [19]. Enfin, on peut souligner que d’autres rôles protecteurs ont été attribués au 2MeCE (rôle protecteur dans les cellules endothéliales, probablement anti-oxydant).
Devant ces effets opposés des catéchols, il avait été évoqué que le rapport des métabolites soit plus important que leurs quantités intrinsèques. Le rapport 4CE/2CE semble en effet augmenter dans les extraits ou les microsomes de cellules tumorales mammaires [16].
Comme la concentration de ces deux catéchols est étroitement liée à l’expression des enzymes impliquées dans leur synthèse ou dans leur métabolisme, tout facteur susceptible de modifier leur expression pourrait potentiellement jouer un rôle dans les mécanismes génotoxiques. C’est le cas de plusieurs facteurs dépendants de l’environnement. Ainsi, l’E2 lui-même, est capable de multiplier par 6 le rapport 4CE/2CE dans le rein de hamster doré [3]. Les xénobiotiques comme la dioxine et les xéno-œstrogènes (molécules polluantes présentant une activité œstrogénique) pourraient influencer également ce rapport. En effet, la dioxine -qui est un ligand du récepteur Ah (ou AhR pour aryl hydrocarbon receptor) - induit (comme les autres ligands du AhR) une augmentation de l’expression des CYP1A et 1B [20] dans plusieurs lignées tumorales mammaires MCF-7 et T47D (Figure 2). Cet effet est dû à l’activation du récepteur Ah qui stimule la transcription des gènes codant pour ces enzymes. Toutefois, nous avons montré qu’un traitement simultané par des pesticides inhibe partiellement l’augmentation d’expression du CYP1A1 sans affecter celle du CYP1B1 [21]. Le mécanisme d’action n’est pas encore connu. Cependant, des pesticides comme l’endosulfan activent probablement un ou plusieurs récepteurs nucléaires (récepteurs des œstrogènes, PXR) qui pourraient être impliqués dans ce mécanisme de régulation différentielle. Le déséquilibre entre les niveaux d’expression de ces CYP pourrait favoriser la synthèse de 4CE par rapport à celle de 2CE et ainsi avoir des effets délétères. Cela permet de rendre compte de la toxicité particulière d’un mélange de polluants comme la dioxine et les pesticides. Cette inhibition de l’expression du CYP1A1 est aussi constatée en présence d’E2, l’œstrogène naturel. Ce résultat pourrait expliquer pourquoi une ovariectomie, dont une des conséquences principales est la chute des niveaux d’œstrogènes, diminue l’incidence des cancers mammaires chez les rongeurs exposés à la dioxine [22,23].
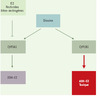 |
Figure 2. Influence de certains xénobiotiques sur l’expression de deux cytochromes impliqués dans le métabolisme des catéchols. Les cytochromes P450 1A1 et 1B1 présentent une spécificité d’hydroxylation de l’E2. Le niveau d’expression de ces enzymes est inductible par la dioxine et les hydrocarbures aromatiques. En revanche, d’autres polluants environnementaux comme certains pesticides ou des molécules endogènes comme E2, inhibent l’expression du CYP1A1. La présence de dioxine et de xéno-œstrogènes conduit à une diminution d’expression du CYP1A1 sans affecter le CYP1B1. Ceci pourrait conduire à un déséquilibre de production de catéchols en faveur du 4OH-E2 et donc à une génotoxicité plus forte. |
Conclusions
Il apparaît donc que les œstrogènes, en particulier l’œstradiol, pourraient jouer un rôle dans le développement de certains cancers en produisant des métabolites génotoxiques.
Cependant, l’importance des catéchols en physiopathologie est encore discutée en raison des quantités élevées d’œstrogènes nécessaires à leur synthèse. Toutefois, certaines activités de détoxication peuvent être très faibles chez certains individus, ce qui favoriserait l’accumulation de catéchols dans les tissus. Par ailleurs, une synthèse locale d’œstrogènes a été détectée dans certains tissus (tissu adipeux, sein) [24], ce qui augmenterait donc les quantités locales de substrats. Enfin, de faibles quantités de catéchols sont nécessaires pour la production de quinones, molécules capables de produire des dérivés réactifs de l’oxygène. On peut également souligner qu’un autre dérivé des œstrogènes, le 16 α-hydroxyestrone, pourrait jouer un rôle important dans la tumorigénèse mammaire [25] bien que le 4CE; semble être le toxique principal.
Si l’hypothèse de la génotoxicité des métabolites des œstrogènes se confirme (Figure 3), ce mécanisme pathologique dépendrait donc, pour chaque enzyme précédemment décrite, de très nombreux paramètres (niveau d’expression, concentration locale en œstrogènes, affinité pour leurs substrats, activité catalyltique, compétition allostérique ou non, …), ce qui expliquerait la difficulté d’isoler un agent étiologique majeur. Des études sont en cours pour établir des relations potentielles entre les polymorphismes du CYP1B1 et les cancers hormono-dépendants
 |
Figure 3. Modèle impliquant les œstrogènes dans la tumorigenèse des tissus. Les œstrogènes agissent selon deux voies ; une première voie, promotrice, impliquant le récepteur des œstrogènes et une augmentation de la croissance des cellules exposées, et, une deuxième voie, “initiatrice”, impliquant les métabolites toxiques de l’E2. Ces composés peuvent être éliminés de l’organisme ou endommager les structures cellulaires, ce qui augmente les risques de tumorigenèse. |
Enfin, ce mécanisme génotoxique n’exclut en rien le rôle des œstrogènes dans la croissance tumorale (Figure 3), et ces deux mécanismes pourraient agir de concert pour stimuler à la fois la prolifération et l’accumulation de mutations .
Références
- Martucci CP, Fishman J. P450 enzymes of estrogen metabolism. Pharmacol Ther 1993; 57 : 237–57. [Google Scholar]
- Ozawa Y Higashiyama T, Shimizu Y, Yarborough C. Multiple functions of aro-matase and the active site structure; aromatase is the placental estrogen 2-hydroxylase. J Steroid Biochem Mol Biol 1993; 44 : 469–80. [Google Scholar]
- Weisz J, Bui QD, Roy D, Liehr JG. Elevated 4-hydroxylation of estradiol by hamster kidney microsomes: a potential pathway of metabolic activation of estrogens. Endocrinology 1992; 131 : 655–61. [Google Scholar]
- Hayes CL, Spink DC, Spink BC, Cao JQ, Walker NJ, Sutter TR. 17 beta-estradiol hydroxylation catalyzed by human cytochrome P450 1B1. Proc Natl Acad Sci USA 1996; 93: 9776–81. [Google Scholar]
- Stresser DM, Kupfer D. Catalytic characteristics of CYP3A4: requirement for a phenolic function in ortho hydroxylation of estradiol and mono-Odemethylated methoxychlor. Biochemistry 1997; 36 : 2203–10. [Google Scholar]
- Cavalieri E, Frenkel K, Liehr JG, Rogan E, Roy D. Estrogens as endogenous genotoxic agents-DNA adducts and mutations.J Natl Cancer Inst Monogr 2000; 27 : 75–93. [Google Scholar]
- Yager JD. Endogenous estrogens as carcinogens through metabolic activation. J Natl Cancer Inst Monogr 2000; 27: 67–73. [Google Scholar]
- Liehr JG, Roy D, Ari-Ulubelen A, Bui QD, Weisz J, Strobel HW. Effect of chronic estrogen treatment of Syrian hamsters on microsomal enzymes mediating formation of catecholestrogens and their redox cycling: implications for carcinoge-nesis. J Steroid Biochem 1990; 35 : 555–60. [Google Scholar]
- Yager JD, Liehr JG. Molecular mechanisms of estrogen carcinogenesis. Annu Rev Pharmacol Toxicol 1996; 36 : 203–32. [Google Scholar]
- Wang MY, Liehr JG. Identification of fatty acid hydroperoxide cofactors in the cytochrome P450-mediated oxidation of estrogens to quinone metabolites. Rôle and balance of lipid peroxides during estrogen-induced carcinogenesis.J Biol Chem 1994; 269, 284–91. [Google Scholar]
- Roy D, Strobel HW, Liehr JG. Cytochrome b5-mediated redox cycling of estrogen. Arch Biochem Biophys 1991; 285; 331–8. [Google Scholar]
- Li JJ, Li SA. Estrogen carcinogenesis in Syrian hamster tissues: role of metabolism. Fed Proc 1987; 46 : 1858–63. [Google Scholar]
- Liehr JG, Fang WF, Sirbasku DA, Ari-Ulubelen A. Carcinogenicity of catechol estrogens in Syrian hamsters. J Steroid Biochem 1986; 24 : 353–6. [Google Scholar]
- Cavalieri EL, Stack DE, Devanesan PD, et al. Molecular origin of cancer: catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc Natl Acad Sci USA 1997; 94 : 10937–42. [Google Scholar]
- Liehr JG, Ricci MJ, Jefcoate CR, Hannigan EV, Hokanson JA, Zhu BT. 4-Hydroxylation of estradiol by human uterine myometrium and myoma microsomes: implications for the mechanism of uterine tumorigenesis. Proc Natl Acad Sci USA 1995; 92 : 9220–4. [Google Scholar]
- Liehr JG, Ricci MJ. 4-Hydroxylation of estrogens as marker of human mammary tumors.Proc Natl Acad Sci USA 1996; 93 : 3294–6. [Google Scholar]
- Han X, Liehr JG. Microsomemediated 8-hydroxylation of guanine bases of DNA by steroid estrogens: correlation of DNA damage by free radicals with metabolic activation to quinones. Carcinogenesis 1995; 16 : 2571–4. [Google Scholar]
- Creveling, C.R. Catechol-omethyltransferase: factors relating to the carcinogenic potential of catecholestrogens. Polycyclic Aromatic Compounds 1994; 125. [Google Scholar]
- Roy D, Weisz J, Liehr JG. The O-methylation of 4-hydroxyestradiol is inhibited by 2-hydroxyestradiol: implications for estrogeninduced carcinogenesis. Carcinogenesis 1990; 11 : 459–62. [Google Scholar]
- Lesca P,Pineau T. Toxicité de la dioxine : rÔle des protéines “PAS”. Med Sci 1999; 15 : 1379–87 [Google Scholar]
- Coumoul X, Diry M, Robillot C, Barouki R. Differential regulation of cytochrome P450 1A1 and 1B1 by a combination of dioxin and pesticides in the breast tumor cell line MCF-7. Cancer Res 2001; 61 : 3942–8. [Google Scholar]
- Lucier GW, Tritscher A, Goldsworthy T, et al. Ovarian hormones enhance 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated increases in cell proliferation and preneoplastic foci in a two-stage model for rat hepatocarcinogenesis. Cancer Res 1991; 51 : 1391–7. [Google Scholar]
- Tritscher AM, Seacat AM, Yager JD, et al. Increased oxidative DNA damage in livers of 2,3,7,8-tetrachlorodibenzo-p-dioxin treated intact but not ovariectomi-zed rats. Cancer Lett 1996; 98 : 219–25. [Google Scholar]
- Jefcoate CR, Liehr JG, Santen RJ, et al. Tissuespecific synthesis and oxidative metabolism of estrogens.J Natl Cancer Inst Monogr 2000; 27 : 95–112. [Google Scholar]
- Bradlow HL, Hershcopf R, Martucci C, Fishman J. 16 alpha-hydroxylation of estradiol: a possible risk marker for breast cancer. Ann NY Acad Sci 1986; 464: 138–51. [Google Scholar]
Liste des figures
|
Figure 1. Voies métaboliques de l’œstradiol. L’œstradiol (E2, à gauche sur la figure) est le principal œstrogène. Il est métabolisé dans les cellules par différents types de cytochromes P450 (CYP) dont l’expression tissulaire est variable et, dans certains cas, inductible. Les produits de ces réactions sont les catéchols, qui, par action d’autres CYP ou de peroxydases, peuvent former des semi-quinones et des quinones. La formation des quinones peut être non enzymatique et aboutir à la formation d’ion superoxyde O2°-, dérivé réactif de l’oxygène très toxique. La présence de la P450 réductase permet un cycle entre semi-quinones et quinones qui, bien qu’elles soient présentes en petite quantité, conduit souvent à la formation de dérivés réactifs de l’oxygène. Les quinones sont les produits terminaux de la réaction et présentent une affinité pour l’ADN (toxique dans le cas de la E2-3,4-quinone). Différentes enzymes de détoxication (COMT, …) interviennent tout au long de la voie métabolique avec, pour rôle principal, d’inactiver l’une des fonctions hydroxyles des dérivés de l’œstradiol. Sont représentées respectivement en vert et en rouge les voies ou molécules dont le rôle protecteur ou toxique est avéré (inspirée de [24]). |
| Dans le texte | |
|
Figure 2. Influence de certains xénobiotiques sur l’expression de deux cytochromes impliqués dans le métabolisme des catéchols. Les cytochromes P450 1A1 et 1B1 présentent une spécificité d’hydroxylation de l’E2. Le niveau d’expression de ces enzymes est inductible par la dioxine et les hydrocarbures aromatiques. En revanche, d’autres polluants environnementaux comme certains pesticides ou des molécules endogènes comme E2, inhibent l’expression du CYP1A1. La présence de dioxine et de xéno-œstrogènes conduit à une diminution d’expression du CYP1A1 sans affecter le CYP1B1. Ceci pourrait conduire à un déséquilibre de production de catéchols en faveur du 4OH-E2 et donc à une génotoxicité plus forte. |
| Dans le texte | |
|
Figure 3. Modèle impliquant les œstrogènes dans la tumorigenèse des tissus. Les œstrogènes agissent selon deux voies ; une première voie, promotrice, impliquant le récepteur des œstrogènes et une augmentation de la croissance des cellules exposées, et, une deuxième voie, “initiatrice”, impliquant les métabolites toxiques de l’E2. Ces composés peuvent être éliminés de l’organisme ou endommager les structures cellulaires, ce qui augmente les risques de tumorigenèse. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.