")
")
| Issue |
Med Sci (Paris)
Volume 32, Number 2, Février 2016
|
|
|---|---|---|
| Page(s) | 167 - 174 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20163202010 | |
| Published online | 2 mars 2016 | |
Intégration des vecteurs AAV et mutagenèse insertionnelle
Integration of AAV vectors and insertional mutagenesis
Centre international de recherche en infectiologie (CIRI), Inserm U1111, CNRS UMR5308, équipe NucléoVir, École normale supérieure de Lyon, 46, allée d’Italie, 69007 Lyon, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
**
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les vecteurs adéno-associés recombinants (AAVr) sont des outils particulièrement efficaces pour le transfert de gènes in vivo. De ce fait, un grand nombre d’essais précliniques et cliniques de thérapies géniques utilisent ces vecteurs pour le traitement de maladies héréditaires ou acquises. Les vecteurs AAVr sont capables de persister à long terme dans les tissus transduits, sous une forme transcriptionnellement active et majoritairement extra-chromosomique (ou épisomale). Cependant, plusieurs études ont montré qu’une fraction non négligeable du génome AAVr pouvait également s’intégrer de façon non spécifique dans le génome de la cellule hôte, soulevant ainsi la possibilité d’évènements de mutagenèse insertionnelle. Cette revue de synthèse se propose de faire le point sur l’état de nos connaissances quant à l’intégration du virus AAV sauvage et des vecteurs AAV recombinants ainsi que de souligner quelles sont les questions majeures qui restent pour l’instant sans réponse.
Abstract
Recombinant AAV vectors (rAAV) are considered as very efficient tools for in vivo gene transfer. Accordingly, several preclinical and clinical gene therapy trials use these vectors to treat inherited and acquired diseases. rAAV vectors possess the capacity to persist for a long term in the transduced tissue in a transcriptionally active, extra-chromosomal (episomal) form. However, many studies have shown that a significant fraction of the rAAV genomes can also nonspecifically integrate into the host cell genome thus raising the possibility of insertional mutagenesis events. This review summarizes the current knowledge on integration of wild type and rAAV genomes and highlights the major questions which remain unresolved.
© 2016 médecine/sciences – Inserm
Les vecteurs adéno-associés recombinants, ou AAVr, occupent depuis plusieurs années une position privilégiée sur le podium des vecteurs viraux utilisés pour le transfert de gène, en particulier en thérapie génique. Développés vers la fin des années 1980 à partir du virus adéno-associé (AAV), un parvovirus1 humain inconnu de la plupart des virologistes [1], les AAVr ont depuis investi de façon importante un grand nombre de laboratoires impliqués dans le développement de protocoles de thérapie génique pour le traitement de maladies héréditaires ou acquises à l’exclusion de pathologies tumorales. En effet, les vecteurs AAVr ont immédiatement séduit les chercheurs grâce à leur capacité à infecter de façon efficace un grand nombre de tissus de l’organisme dont, entre autres, le foie, le muscle et les systèmes nerveux central et périphérique [2]. Contrairement à ce qui est observé avec d’autres vecteurs viraux, comme ceux qui dérivent de l’adénovirus, les particules d’AAV n’induisent que de faibles réponses immunitaires innées, ce qui permet de limiter la réponse immunitaire adaptative dirigée contre les cellules ciblées par le vecteur [3]. Gage suprême d’une sécurité optimale, les vecteurs AAVr sont dérivés d’un virus initialement défini comme non pathogène chez l’homme, espèce animale qu’il infecte au même titre que bien d’autres [1]. L’absence d’effets adverses constatée jusqu’à présent sur l’ensemble des patients auxquels ont été administrées des doses importantes de vecteurs AAVr constituerait une preuve supplémentaire de l’innocuité de ces vecteurs pour l’organisme [2]. Or, deux articles récents semblent remettre en cause cette vision en replaçant sur le devant de la scène un rôle oncogène potentiel du virus AAV sauvage, et des vecteurs AAVr, dû à leur intégration dans le génome de la cellule cible [4, 5]. Peut-on relier ces deux observations et, d’une façon générale, peut-on comprendre le comportement des vecteurs AAVr en se basant sur les propriétés intégratives du virus AAV sauvage ?
Le virus AAV sauvage et sa capacité d’intégration site-spécifique
Le virus AAV sauvage est composé d’une capside non enveloppée de 20 nm de diamètre contenant une molécule d’ADN simple brin de 4,7 kb (Figure 1). Le génome viral contient trois cadres de lecture ouverts qui codent quatre protéines régulatrices Rep, trois protéines structurales VP (ou Cap) et une protéine accessoire AAP (assembly-activating protein). Le génome est encadré par des petites répétitions terminales (ITR, inverted terminal repeats) de 145 pb qui forment des structures palindromiques complexes essentielles pour la réplication du génome viral et son encapsidation. Identifié au départ comme un virus satellite, c’est-à-dire dépendant d’un autre virus pour sa réplication (d’où sa classification comme Dépendovirus au sein de la famille des Parvovirus), le virus AAV a défrayé la chronique scientifique des années 1990 en montrant qu’il était capable de s’intégrer de façon site-spécifique dans un locus du chromosome 19 humain, appelé depuis AAVS1 (adeno-associated virus integration site 1) [6–8]. Les études conduites pour comprendre le mécanisme d’intégration ont révélé que celui-ci survenait grâce à la présence, sur le site AAVS1, de deux motifs également présents sur les ITR virales : un site de fixation des protéines Rep (RBS, Rep binding site) et un site de clivage (trs, terminal resolution site) cible de ces mêmes protéines. La présence de ces deux motifs, sur les ITR de l’AAV et sur le site AAVS1, permettrait aux protéines Rep de réaliser un pontage entre l’ADN viral et l’ADN chromosomique (Figure 2) [9]. L’intégration du génome viral surviendrait à la suite d’un clivage de l’ADN cellulaire réalisé par les protéines Rep et grâce à l’intervention de facteurs cellulaires de réparation de l’ADN [10]. Nos études récentes sur l’intégration du virus AAV sauvage indiquent que le complexe formé par l’assemblage de Mre11, Rad50 et Nbs1 (Mre11-Rad50-Nbs1, MRN complex) qui intervient dans les processus de réparation de l’ADN, est essentiel pour permettre une intégration du génome viral dans le locus AAVS1 [11]. Ce résultat, et d’autres qui montrent une implication de certains facteurs de la voie de réparation non homologue (NHEJ) [12–14], pointe l’attention vers des mécanismes de réparation qui feraient intervenir des voies de réparation de type NHEJ en utilisant, éventuellement, des zones de micro-homologies. En effet, l’intégration de l’AAV sauvage peut avoir lieu dans une zone très étendue, en amont et en aval du site AAVS1, et entraîner des modifications profondes du génome cellulaire (délétions, duplications) ainsi que des délétions des extrémités du génome viral. Il est ainsi impossible de connaître, a priori, les séquences virales présentes au niveau des jonctions avec l’ADN cellulaire, contrairement aux rétrovirus ou aux lentivirus pour lesquels les séquences des jonctions virales peuvent être prédites.
 |
Figure 1. Génomes de l’AAV sauvage et recombinant. Le génome de l’AAV de sérotype 2, représenté ici, contient trois promoteurs (p5, p19 et p40) et un signal de polyadénylation (polyA). Il est encadré par deux répétitions terminales, les ITR (pour inverted terminal repeats) qui sont des régions essentielles pour la réplication et l’encapsidation du génome viral. Le génome sauvage code 8 protéines : les protéines régulatrices Rep (gène rep), les protéines structurales VP (gène cap), et la protéine chaperonne d’assemblage AAP. Le génome du vecteur AAV recombinant est obtenu en remplaçant les gènes viraux par une cassette d’expression du transgène qui contient, au minimum, un promoteur, un ADN complémentaire (ADNc) et un signal de polyA. Les ITR sont donc les seules séquences d’origine virale présentes dans le vecteur AAVr. Ces deux types de génomes sont encapsidés, sous la forme d’une molécule d’ADN simple brin, dans une capside virale de 20 nm de diamètre composée des 3 protéines VP formant une particule d’AAV sauvage ou d’AAVr. |
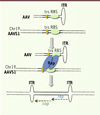 |
Figure 2. Modèle théorique d’intégration du génome de l’AAV sauvage dans le locus AAVS1. L’ITR (inverted terminal repeats) de l’AAV et le locus AAVS1 du génome cellulaire partagent un site de fixation pour les protéines Rep (ou RBS [Rep binding site]) et un site de clivage par ces mêmes protéines (ou trs, terminal resolution site). Selon le modèle d’intégration, les protéines virales Rep seraient capables de réaliser un pontage entre le génome viral et le génome cellulaire via la reconnaissance du site RBS. La fixation de ces protéines au RBS entraîne un clivage de l’ADN cellulaire et viral au niveau du site trs. Le génome viral s’intégrerait alors via un mécanisme encore mal élucidé impliquant plusieurs facteurs de réparation de l’ADN ainsi que des polymérases cellulaires. Des travaux récents ont montré que l’intégration du génome de l’AAV survenait majoritairement en orientation inverse. Il est important aussi de noter que l’intégration virale peut survenir dans une région chromosomique très large et de façon préférentielle en amont du site RBS. Il en résulte, dans la plupart des cas, une délétion partielle ou complète des ITR ainsi que des délétions et/ou des réarrangements des séquences cellulaires adjacentes. Des sites RBS consensus et des sites trs cryptiques ont été également retrouvés en grand nombre dans d’autres régions chromosomiques, ce qui explique la capacité du virus à cibler différents locus cellulaires (voir Figure 3). |
Bien que considérée comme l’une des propriétés les plus originales de l’AAV, la capacité du virus à s’intégrer de façon spécifique et quasi exclusive dans le site AAVS1 a été ensuite remise en cause par les travaux réalisés dans des cellules en culture ou in vivo. Il apparaît, premièrement, que la capacité de l’AAV à s’intégrer semble être largement favorisée par l’état prolifératif des cellules. En effet, la plupart des études ont été réalisées sur des lignées cellulaires transformées, comme les cellules HeLa2 infectées avec des doses importantes d’AAV et les rares analyses in vitro réalisées en utilisant des cellules primaires, ou au repos, suggèrent un niveau et une cinétique d’intégration du génome viral extrêmement réduits [10]. Par ailleurs, la recherche de séquences d’AAV in vivo, dans des tissus humains provenant de donneurs sains tels que les amygdales, la rate, le foie et le muscle, suggère une persistance dans ces tissus, majoritairement sous la forme d’épisomes3 double brin circulaires [15–17]. Dans ces études, seuls quelques rares événements d’intégration (moins de 5 %) ont été détectés mais pas dans le site AAVS1 et, pour la plupart, dans des tissus co-infectés avec de l’adénovirus, comme c’est le cas des amygdales. D’autres analyses, réalisées sur des tissus sains de primates humain et non humain, ont confirmé la présence de génomes AAV dans une grande majorité de tissus et en particulier le foie et la rate. Dans ces tissus, le génome viral est présent sous une forme majoritairement épisomale et seuls quelques événements d’intégrations ont pu être détectés dans des tissus de singes comportant un grand nombre de copies d’AAV par cellule (supérieur à 20) [18, 19]. Des études récentes, réalisées en utilisant des techniques puissantes pour isoler des jonctions entre l’AAV et l’ADN chromosomique, suivies d’un séquençage à haut débit, ont montré, deuxièmement, que lorsqu’il s’intègre, l’AAV ne cible pas exclusivement le chromosome 19 mais aussi un grand nombre d’autres sites chromosomiques (Figure 3A) [20–22]. Cela s’explique par la présence dans le génome cellulaire de nombreux sites RBS reconnus par les protéines Rep de l’AAV. Le point commun à tous ces événements d’intégration est, dans la majorité des cas, la présence d’une seule copie de l’AAV intacte (à l’exception des délétions au niveau des ITR) en orientation inverse. Au vu de ces résultats, l’AAV apparaît comme un virus qui, dans des cellules au repos, persiste majoritairement sous une forme extra-chromosomique mais dont l’intégration pourrait être induite lors de la mise en cycle des cellules ou d’autres modifications métaboliques (Tableau I). Ce scénario est en accord avec le fait que, contrairement à d’autres virus à ADN à persistance épisomale (comme par exemple le virus d’Epstein-Barr, EBV), l’AAV n’est pas en mesure de répliquer son génome épisomal en même temps que l’ADN cellulaire. La division cellulaire entraîne donc une perte par dilution des épisomes d’AAV sauf s’il y a intégration, mécanisme qui permettait au virus de se maintenir dans la cellule cible.
 |
Figure 3. Distribution du nombre d’événements d’intégration de l’AAV sauvage et recombinant par chromosome. Le nombre d’intégrations du génome AAV sauvage ( A ) ou recombinant ( B ) a été mesuré après infection de fibroblastes primaires humains. Il est exprimé en nombre d’événements d’intégration détectés toutes les 10 mégabases (Mb) d’ADN (© Daniela Hüser et Regine Heilbronn, institut de virologie, université de médecine de la Charité, Berlin, Allemagne). |
Résumé du mode de persistance des génomes AAV sauvage et recombinant.
Comme souligné dans l’introduction, malgré sa forte prévalence chez l’homme et l’animal, dans un grand nombre de tissus, l’AAV sauvage a été considéré jusqu’à présent comme un virus non pathogène, voire même comme un virus potentiellement bénéfique pour l’organisme qu’il infecte. En effet, les données moléculaires indiquent que l’AAV peut fortement inhiber plusieurs virus pathogènes qui l’aident à se répliquer tels que l’adénovirus, le virus de l’herpès simplex de type 1 ou le papillomavirus [1]. Cependant, un article très récent remet en question cette notion en montrant la présence de portions d’AAV intégrées dans le génome cellulaire provenant de 11 prélèvements d’hépatocarcinome (HCC) humain sur un total de 193 étudiés [5]. Les auteurs démontrent que dans ces 11 cas d’HCC, l’AAV est intégré dans un site unique au sein de la tumeur, dans des gènes déjà connus pour leur rôle dans le développement de ce carcinome, tels que le gène codant la télomérase (TERT) ou la cycline E1 (CCNE1). De façon surprenante, dans la majorité de ces 11 cas, seule une portion très restreinte du génome AAV, entre 200 et 400 pb, et couvrant l’ITR 3’ est retrouvée intégrée. Des analyses moléculaires montrent que l’intégration de ces portions d’ITR conduit à une surexpression des gènes qui sont ciblés par le virus.
Dans l’ensemble, cette étude soulève la question du rôle oncogène potentiel de l’AAV sauvage après son intégration et, en particulier, à la suite d’une transactivation des gènes cellulaires médiée par la région ITR du virus. Cependant, le faible nombre d’événements d’intégration retrouvés ainsi que la longueur et l’orientation des génomes AAV intégrés posent de nombreuses questions quant aux mécanismes et aux causes de ces intégrations. Qui plus est, le mécanisme responsable de la surexpression des gènes ciblés reste à élucider, bien que l’ITR puisse avoir un rôle de promoteur faible [23]. D’autres études sont donc nécessaires avant de conclure quant à un rôle oncogène de l’AAV sauvage.
Les vecteurs AAVr : persistance épisomale ou intégration ?
La caractérisation initiale, dans les années 1980, de l’AAV comme un virus capable de s’intégrer de façon site-spécifique a constitué un point d’attraction majeur pour son utilisation comme vecteur recombinant. En effet, déjà à cette époque la possibilité de cibler le vecteur dans un locus cellulaire précis apparaissait comme un objectif désirable, comme en témoigne l’essor, dans les années suivantes, des technologies visant à obtenir un ciblage génique spécifique de type Zinc-Finger, TALEN (transcription activator-like effector nuclease) ou plus récemment Crispr-Cas9 [24–26, 46] (→). En contrôlant le site d’intégration d’un vecteur viral dans le génome cellulaire, il deviendrait ainsi possible de limiter, voire de réduire, les risques d’oncogenèse dus à une dérégulation de gènes cellulaires situés à proximité du site d’intégration, comme cela a été observé avec les vecteurs rétroviraux et lentiviraux (pour une revue traitant des problèmes de mutagénèse avec ce type de vecteurs, voir [27, 28]).
(→) Voir la Synthèse de J.P. Tremblay, m/s n° 11, novembre 2015, page 1014
Cependant, l’analyse du mécanisme d’intégration du virus AAV sauvage a très rapidement permis d’en déduire que les vecteurs AAVr ne pouvaient plus s’intégrer de façon efficace dans le site AAVS1 ou dans les autres sites chromosomiques ciblés par l’AAV sauvage, et ceci pour une raison simple : les vecteurs AAVr n’expriment pas les protéines virales Rep qui sont essentielles à l’intégration du virus dans ces régions (Figure 1). Qu’en est-il alors de leur devenir une fois dans le noyau des cellules infectées ? Dès leur première utilisation in vivo comme vecteur de transfert de gène, en particulier chez la souris, les vecteurs AAVr ont surpris les chercheurs par leur capacité à induire une expression stable du transgène qui leur était associé. De façon inattendue, la cinétique d’expression de ces vecteurs in vivo montre, dans la plupart des cas, une augmentation progressive du niveau d’expression du transgène au cours des deux premières semaines après l’injection, suivie d’une phase de stabilisation. En l’absence de réponses immunitaires dirigées contre le produit du transgène, celui-ci peut donc s’exprimer de façon stable pendant toute la vie de l’animal. Cette constatation, d’abord réalisée chez des souris, a été également vérifiée par la suite dans des modèles animaux de plus grande taille ainsi que lors d’essais cliniques réalisés chez l’homme [2]. L’ADN de vecteurs AAVr a ainsi été retrouvé dans des biopsies de muscles provenant de patients chez qui on avait injecté ces vecteurs plus de 4 ans auparavant [29].
Les analyses conduites pour déterminer la forme sous laquelle persistent les vecteurs AAVr ont montré que, dans des tissus comme le muscle ou le foie, leurs génomes persistaient majoritairement sous des formes extra-chromosomiques circulaires double brin parfois constituées de plusieurs molécules de vecteurs reliées entre elles pour former des concatémères4 [30–34]. Comme dans le cas du virus AAV sauvage, les génomes d’AAVr se maintiennent donc dans les cellules au repos majoritairement sous la forme d’épisomes recouverts d’histones, transcriptionnellement actifs, qui peuvent être perdus par dilution lors de la division cellulaire. Ainsi, des expériences réalisées dans des foies de souris en régénération ont permis de montrer la diminution progressive de l’expression du transgène qui est associée à une perte de génomes AAV recombinants dans les hépatocytes qui se divisent [35]. Cependant, leur persistance in vivo sous une forme majoritairement extra-chromosomique, n’excluait pas la possibilité que les vecteurs AAVr soient à même de s’intégrer dans le génome cellulaire, même à un niveau faible. En effet, des expériences réalisées sur des cellules en culture avaient déjà montré que, bien qu’ayant perdu leur capacité d’intégration site-spécifique, les vecteurs AAVr peuvent s’intégrer à un niveau faible, mais détectable, dans des gènes transcriptionnellement actifs [36]. Comme dans le cas de l’AAV sauvage, l’intégration des vecteurs AAVr dans des cellules en culture peut survenir dans plusieurs régions chromosomiques, mais les locus ciblés sont différents de ceux du virus sauvage (Figure 3B) [22]. Il apparaît aussi que les sites d’intégration de l’AAVr varient en fonction du type cellulaire ciblé confirmant la préférence de l’AAVr pour des régions transcriptionnellement actives qui dépendent du tissu d’origine de ces cellules [22]. De même, les expériences réalisées in vivo dans des foies de souris en régénération ont permis de montrer qu’environ 10 % des génomes AAVr étaient effectivement intégrés dans le génome cellulaire [35, 37, 38]. La caractérisation de ces sites d’intégration a confirmé que les génomes AAVr ciblent de façon préférentielle les gènes transcriptionnellement actifs avec une préférence pour les régions adjacentes aux sites d’initiation de la transcription et les îlots CpG5 ; le pourcentage de génomes AAVr intégrés à proximité d’oncogènes cellulaires étant estimé à 3,5 % [38].
Ainsi, comme pour le virus sauvage, une fraction minoritaire mais non négligeable des génomes AAVr peut s’intégrer dans le génome cellulaire et potentiellement perturber l’expression des gènes adjacents.
Utilisation des vecteurs AAVr in vivo et mutagenèse insertionnelle
La majorité des études qui ont exploré un rôle oncogène éventuel des vecteurs AAVr a été réalisée sur le foie, organe cible de nombreuses stratégies et protocoles de thérapie génique, un grand nombre de maladies métaboliques du foie pouvant être potentiellement traitées par ce type de thérapie. Par ailleurs, le foie peut être facilement infecté par les vecteurs AAVr injectés dans la circulation sanguine. Les hépatocytes ainsi infectés peuvent également servir de cellule source pour la production de facteurs solubles dont la synthèse est altérée. C’est le cas par exemple dans les essais cliniques qui ont été réalisés pour l’hémophilie de type B6 dans lesquels le gène fonctionnel codant le facteur IX humain a été empaqueté dans le vecteur AAVr qui l’achemine dans les cellules du foie où le facteur de coagulation est normalement produit. Enfin, comme nous l’avons déjà souligné, le foie constitue un organe particulier grâce à ses capacités de régénération et, du point de vue immunitaire, à son statut d’organe tolérogène.
Plusieurs essais réalisés chez la souris ont permis de suivre l’émergence de tumeurs hépatiques plusieurs mois après injection des vecteurs AAVr. L’utilisation du modèle murin offre l’avantage de pouvoir réaliser ces observations sur un grand nombre d’animaux, en faisant varier la souche de souris, la séquence du vecteur et le sérotype d’AAV. Ainsi, plusieurs études ont démontré que le taux d’émergence de tumeurs hépatiques chez les souris ayant reçu des vecteurs AAVr n’était pas supérieur à celui observé dans des souris contrôles, non traitées [39, 40]. Une première étude, très médiatisée, est venue troubler ce tableau rassurant [41]. Dans celle-ci, les chercheurs démontraient la survenue de tumeurs hépatiques dans environ un tiers des souris injectées au stade nouveau-né avec un vecteur AAVr codant une enzyme lysosomale, la β-glucuronidase. L’analyse des sites d’intégration a permis de déterminer que, dans la plupart des tumeurs, le génome des vecteurs était intégré dans un locus précis du chromosome 2 murin, appelé Rian. Ce locus constitue une région chromosomique complexe qui contient de nombreux gènes transcrits en ARN non codants dont des micro-ARN (miARN), des petits ARN nucléolaires (snoARN) et des longs ARN non codants (lncARN) qui régulent l’expression de plusieurs gènes cellulaires. La dérégulation de l’expression de la région homologue chez l’homme a été associée au développement d’hépatocarcinome. Ainsi, l’intégration du vecteur AAVr dans cette région a été proposée comme un mécanisme possible à l’origine du processus tumoral. Dès lors, et malgré bien d’autres études démontrant l’absence d’effet tumorigène, la question du potentiel oncogène des vecteurs AAVr est restée en suspens et est devenue un sujet de controverse.
Une deuxième étude récente vient de remettre un peu d’ordre dans ce tableau en explorant de façon plus extensive l’apparition de tumeurs hépatiques chez la souris et en déterminant quelques paramètres critiques [4]. Dans cette étude, Chandler et al. ont suivi plusieurs cohortes de souris toutes injectées au stade nouveau-né (1 jour après la naissance) par voie intraveineuse avec des vecteurs AAVr codant la méthylmalonyl CoA mutase (Mut), en vue de l’élaboration d’un traitement par thérapie génique de l’acidémie méthylmalonique (MMA), une maladie métabolique qui touche une population de patients pédiatriques. En suivant les animaux pendant 22 mois, les auteurs ont observé un taux de cancer du foie touchant, en moyenne, 80 % des animaux traités alors que ce taux était inférieur à 5 % pour les animaux non traités. Ils ont également déterminé que l’incidence de cancer était indépendante du type de transgène véhiculé par le vecteur AAVr et de la souche de souris. En revanche, elle était strictement corrélée à la dose de vecteur utilisée lors de l’injection qui s’élevait, dans ces essais comme dans bon nombre d’études précliniques et cliniques, à environ 1011 à 1012 particules de vecteur/kg. Une analyse détaillée des sites d’intégration a pu être réalisée grâce à une méthode de capture des jonctions entre l’ADN cellulaire et viral suivie d’un séquençage à haut débit. Cette analyse a montré que l’intégration des vecteurs AAVr survient dans un nombre limité de gènes, la majorité d’entre eux étant fortement exprimée dans le foie. C’est le cas par exemple du gène de l’albumine et de celui de l’alpha-fœtoprotéine qui cumulaient à eux seuls respectivement 80 % et 30 % des événements d’intégration uniques, et cela aussi bien dans des cellules tumorales que dans des cellules non transformées. Ce résultat vient ainsi confirmer les études précédentes montrant une intégration préférentielle des vecteurs AAVr dans des régions transcriptionnellement actives sans pour autant révéler une quelconque relation de causalité entre l’intégration dans ces régions et l’émergence de la tumeur. De façon très intéressante, parmi l’ensemble des locus ciblés par l’AAVr, seul le locus Rian montrait une fréquence d’intégration significativement supérieure dans les cellules tumorales par rapport à celle que l’on retrouve dans des hépatocytes normaux localisés autour de la tumeur. En outre, une majorité des intégrations était retrouvée dans le gène Mir341, qui est inclus dans le locus Rian et code le miARN 341. Ainsi, cette analyse vient conforter, avec une force statistique bien supérieure, le résultat précédemment décrit par Donsante et al. qui avaient mis en évidence le ciblage de ce même locus par les vecteurs AAVr et son association avec des événements tumoraux [41].
Mais comment un événement d’intégration dans cette région peut-il entraîner la progression vers un état tumoral ? C’est là que l’histoire rejoint très probablement celle des vecteurs rétroviraux et lentiviraux pour lesquels des événements de mutagenèse insertionnelle ont été reliés à une activation transcriptionnelle des gènes adjacents aux sites d’intégration [27, 28]. En effet, une analyse comparative des ARN transcrits par les hépatocytes des souris ayant reçu l’AAVr a permis de mettre en évidence une surexpression globale des ARN codants et non codants situés à proximité du locus Rian et cela exclusivement dans les hépatocytes tumoraux. Parmi ces gènes se retrouvaient des marqueurs d’hépatocarcinome ainsi qu’un élément transposable, désigné Rtl1 (retrotransposon-like 1), déjà associé à la formation de tumeurs du foie chez la souris [42]. Des analyses similaires, réalisées avec d’autres vecteurs AAVr contenant différents types de promoteurs, ont montré que l’incidence d’hépatocarcinome dépendait, en fait, de la séquence promotrice qui était utilisée dans les constructions. Plus spécifiquement, de fortes incidences tumorales ont été observées avec des promoteurs forts d’origine virale provenant du cytomégalovirus, ou cellulaires comme celui de l’α-1-microglobuline. En revanche, l’utilisation d’autres types de promoteurs plus faibles, comme celui du gène de l’α-1-antitrypsine humaine, ne conduisait pas à une amplification de l’émergence de tumeurs et cela malgré une intégration détectable dans le locus Rian, montrant que l’insertion de ce type de vecteur dans le génome cellulaire ne dérégule pas l’expression des gènes cellulaires de ce locus, contrairement à ce qui avait été constaté avec les autres constructions. Ainsi, comme dans le cas des vecteurs rétroviraux et lentiviraux, le type de promoteur utilisé pour exprimer le transgène exerce un rôle déterminant dans la dérégulation des gènes cellulaires localisés à proximité du site d’intégration. Ces études indiquent aussi que les ITR virales, présentes dans tous les génomes AAV recombinants, quel que soit le type de promoteur ou de transgène véhiculé, n’auraient pas de rôle dans les événements de mutagenèse insertionnelle observés chez la souris. Cela contraste avec l’observation récente de Nault et al., réalisée sur des prélèvements d’hépatocarcinomes humains, qui suggère un rôle de l’ITR dans la transactivation des gènes cellulaires ciblés par l’intégration du génome AAV sauvage [5]. Comment alors expliquer cette différence de comportement ? Il est possible que le modèle murin ne suffise pas à rendre compte de l’ensemble des événements pouvant survenir dans les cellules humaines. Il est aussi envisageable que l’activité transactivatrice des ITR ne puisse pas s’exercer dans les vecteurs AAV recombinants à cause de la présence d’un promoteur transcriptionnellement actif. Quelle que soit l’explication, il est probable que de nombreuses analyses, réalisées à partir d’échantillons de foie provenant d’études précliniques et cliniques, seront conduites pour déterminer si les vecteurs AAV recombinants peuvent également s’intégrer à proximité d’oncogènes connus et si des formes réduites, composées exclusivement de portions d’ITR, peuvent être retrouvées.
Conclusion et pistes de recherche
De toute évidence, les résultats présentés dans l’article de Chandler et al. apportent un peu de clarté dans un domaine de recherche encore mal exploré. Ils confirment, tout d’abord, la possibilité d’intégration des vecteurs AAVr dans le foie murin et montrent que le niveau d’expression des gènes cellulaires constitue un facteur décisif pour orienter leur intégration. L’intégration des vecteurs AAVr est vraisemblablement favorisée, comme pour le virus sauvage, par un état de chromatine « ouverte » généralement associé à des régions actives du point de vue transcriptionnel. Il est aussi possible que, comme observé dans des cellules en culture, l’intégration de l’AAVr advienne de façon préférentielle dans des zones de cassure de l’ADN cellulaire qui seraient alors « réparées » en y intégrant des génomes viraux [43]. La deuxième conclusion essentielle de cette étude est que la séquence du vecteur, plus que la nature de sa capside, constitue un critère décisif pour les conséquences des événements d’intégration. En particulier, la « force » des régions promotrices et leur capacité à transactiver des gènes cellulaires à distance, constitue un facteur déterminant pour la progression tumorale. Il reste à déterminer si, comme dans le cas du virus sauvage, les régions ITR pourraient, dans certaines configurations, activer la transcription de gènes adjacents. Il est donc nécessaire de disposer, comme dans le cas de vecteurs rétroviraux et lentiviraux, de modèles animaux pertinents afin d’évaluer précisément ce risque avant de procéder à des essais cliniques. Une fois ces conclusions établies, que reste-t-il à comprendre et à réaliser pour réduire ultérieurement le risque de carcinogenèse insertionnelle des vecteurs AAVr ?
Très clairement, compte tenu du scénario décrit ci-dessus, on comprend aisément que l’intégration des vecteurs AAVr dépende des doses injectées. De ce point de vue, il apparaît essentiel de réduire les doses de vecteurs tout en conservant une efficacité thérapeutique satisfaisante. La génération de nouvelles capsides d’AAV, génétiquement modifiées et sélectionnées en fonction de leur efficacité de transduction, devrait permettre dans un futur proche d’atteindre au moins en partie cet objectif [44]. Mais au-delà des solutions fournies par l’ingénierie biotechnologique, il est tout aussi essentiel d’identifier la lumière sur les mécanismes impliqués dans l’intégration des vecteurs AAVr qui restent pour le moins obscurs. Tout d’abord, s’il est établi que l’injection des vecteurs AAVr dans le foie des souris nouveau-nés peut conduire à la formation de tumeurs, cela ne semble pas être le cas lorsque les vecteurs sont injectés à des souris plus âgées voire adultes [39, 40, 45]. Cette observation doit être mise en relation avec le fait que le foie, comme tout autre tissu, se développe et augmente de taille pendant les premiers mois du développement extra-utérin de la souris. Les questions qui surgissent sont alors nombreuses : peut-on dire que, comme cela semble être le cas pour l’AAV sauvage, les vecteurs ne s’intégreraient de façon efficace que lorsque les cellules se divisent ? Et quels sont les mécanismes et les facteurs cellulaires mis en jeu ? S’agit-il d’un mécanisme conventionnel d’intégration non homologue (C-NHEJ) ou existe-t-il d’autres processus possibles ? Reste enfin à déterminer si l’intégration des vecteurs AAVr peut également survenir dans d’autres tissus que le foie qui, même dans un organisme adulte, reste un tissu capable d’auto-renouvellement. Cette question n’est pas anodine, car de nombreux essais cliniques utilisent des vecteurs AAVr pour cibler d’autres tissus comme ceux présents dans le cerveau. Peut-on détecter une intégration du vecteur AAVr dans ce type de tissu ? Et si tel n’est pas le cas, quelles en sont les raisons ? Voilà quelques-unes des questions qui restent en suspens et qui font que l’AAV, outre qu’il représente un formidable outil biotechnologique, demeure, comme tout autre virus, un excellent modèle d’étude pour comprendre des mécanismes cellulaires fondamentaux.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Leur nom vient de leur très petite taille (environ 20 nm de diamètre). Ce sont des virus à ADN, non enveloppés, composés d’une capside icosaédrique.
Cellules épithéliales transformées provenant d’une patiente atteinte du cancer du col de l’utérus.
Un épisome est un segment d’ADN circulaire non intégré au génome de la cellule.
Longue molécule d’ADN constituée d’un même monomère répété et formant un multimère.
Les îlots CpG sont des fragments d’ADN riches en guanine (G) et cytosine (C) et dont la proportion en CpG est plus élevée que dans le reste du génome. Ils constituent des sites de méthylation de l’ADN.
L’hémophilie B, qui touche un garçon sur 25 000 naissances, est une maladie génétique liée à un déficit du facteur de coagulation IX.
Références
- Flotte TR, Berns KI. Adeno-associated virus: a ubiquitous commensal of mammals. Hum Gene Ther 2005 ; 16 : 401–407. [CrossRef] [PubMed] [Google Scholar]
- Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev 2011 ; 12 : 341–355. [Google Scholar]
- Rogers GL, Martino AT, Aslanidi GV, et al. Innate immune responses to AAV vectors. Front Microbiol 2011 ; 2 : 194. [CrossRef] [PubMed] [Google Scholar]
- Chandler RJ, LaFave MC, Varshney GK, et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J Clin Invest 2015 ; 125 : 870–880. [CrossRef] [PubMed] [Google Scholar]
- Nault JC, Datta S, Imbeaud S, et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat Genet 2015 ; 47 : 1187–1193. [CrossRef] [PubMed] [Google Scholar]
- Kotin RM, Siniscalco M, Samulski RJ, et al. Site-specific integration by adeno-associated virus. Proc Natl Acad Sci USA 1990 ; 87 : 2211–2215. [CrossRef] [Google Scholar]
- Kotin RM, Linden RM, Berns KI. Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. EMBO J 1992 ; 11 : 5071–5078. [PubMed] [Google Scholar]
- Linden MR, Ward P, Giraud C, et al. Site-specific integration by adeno-associated virus. Proc Natl Acad Sci USA 1996 ; 93 : 11288–11294. [CrossRef] [Google Scholar]
- Weitzman MD, Kyostio SR, Kotin RM, Owens RA. Adeno-associated virus (AAV) Rep proteins mediate complex formation between AAV DNA and its integration site in human DNA. Proc Natl Acad Sci USA 1994 ; 91 : 5808–5812. [CrossRef] [Google Scholar]
- Henckaerts E, Linden RM. Adeno-associated virus: a key to the human genome? Future Virol 2010 ; 5 : 555–574. [CrossRef] [PubMed] [Google Scholar]
- Millet R, Jolinon N, Nguyen XN, et al. Impact of the MRN complex on adeno-associated virus integration and replication during co-infection with herpes simplex virus type 1. J Virol 2015 ; 89 : 6824–6834. [CrossRef] [PubMed] [Google Scholar]
- Daya S, Cortez N, Berns KI. Adeno-associated virus site-specific integration is mediated by proteins of the nonhomologous end-joining pathway. J Virol 2009 ; 83 : 11655–11664. [CrossRef] [PubMed] [Google Scholar]
- Song S, Laipis PJ, Berns KI, Flotte TR. Effect of DNA-dependent protein kinase on the molecular fate of the rAAV2 genome in skeletal muscle. Proc Natl Acad Sci USA 2001 ; 98 : 4084–4088. [CrossRef] [Google Scholar]
- Song S, Lu Y, Choi YK, et al. DNA-dependent PK inhibits adeno-associated virus DNA integration. Proc Natl Acad Sci USA 2004 ; 101 : 2112–2116. [CrossRef] [Google Scholar]
- Schnepp BC, Jensen RL, Chen CL, et al. Characterization of adeno-associated virus genomes isolated from human tissues. J Virol 2005 ; 79 : 14793–14803. [CrossRef] [PubMed] [Google Scholar]
- Schnepp BC, Jensen RL, Clark KR, Johnson PR. Infectious molecular clones of adeno-associated virus isolated directly from human tissues. J Virol 2009 ; 83 : 1456–1464. [CrossRef] [PubMed] [Google Scholar]
- Chen C-L, Jensen RL, Schnepp BC, et al. Molecular characterization of adeno-associated viruses infecting children. J Virol 2005 ; 79 : 14781–14792. [CrossRef] [PubMed] [Google Scholar]
- Gao G, Alvira MR, Somanathan S, et al. Adeno-associated viruses undergo substantial evolution in primates during natural infection. Proc Natl Acad Sci USA 2003 ; 100 : 6081–6086. [CrossRef] [Google Scholar]
- Gao G, Vandenberghe LH, Alvira MR, et al. Clades of adeno-associated viruses are widely disseminated in human tissues. J Virol 2004 ; 78 : 6381–6388. [CrossRef] [PubMed] [Google Scholar]
- Hüser D, Gogol-Doring A, Lutter T, et al. Integration preferences of wildtype AAV-2 for consensus rep-binding sites at numerous loci in the human genome. PLoS Pathog 2010 ; 6 : e1000985. [CrossRef] [PubMed] [Google Scholar]
- Janovitz T, Klein IA, Oliveira T, et al. High-throughput sequencing reveals principles of adeno-associated virus serotype 2 integration. J Virol 2013 ; 87 : 8559–8568. [CrossRef] [PubMed] [Google Scholar]
- Huser D, Gogol-Doring A, Chen W, Heilbronn R. Adeno-associated virus type 2 wild-type and vector-mediated genomic integration profiles of human diploid fibroblasts analyzed by third-generation PacBio DNA sequencing. J Virol 2014 ; 88 : 11253–11263. [CrossRef] [PubMed] [Google Scholar]
- Haberman RP, McCown TJ, Samulski RJ. Novel transcriptional regulatory signals in the adeno-associated virus terminal repeat A/D junction element. J Virol 2000 ; 74 : 8732–8739. [CrossRef] [PubMed] [Google Scholar]
- Dion S, Demattei MV, Renault S. Les domaines à doigts de zinc : vers la modification de la structure et de l’activité des génomes. Med Sci (Paris) 2007 ; 23 : 834–839. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Dupret B, Angrand PO. L’ingénierie des génomes par les TALEN. Med Sci (Paris) 2014 ; 30 : 186–193. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- La Gilgenkrantz H. révolution de CRISPR est en marche. Med Sci (Paris) 2014 ; 30 : 1066–1069. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Schambach A, Zychlinski D, Ehrnstroem B, Baum C. Biosafety features of lentiviral vectors. Hum Gene Ther 2013 ; 24 : 132–142. [CrossRef] [PubMed] [Google Scholar]
- Uren AG, Kool J, Berns A, van Lohuizen M. Retroviral insertional mutagenesis: past, present and future. Oncogene 2005 ; 24 : 7656–7672. [CrossRef] [PubMed] [Google Scholar]
- Jiang H, Pierce GF, Ozelo MC, et al. Evidence of multiyear factor IX expression by AAV-mediated gene transfer to skeletal muscle in an individual with severe hemophilia B. Mol Ther 2006 ; 14 : 452–455. [CrossRef] [PubMed] [Google Scholar]
- Schnepp BC, Clark KR, Klemanski DL, et al. Genetic fate of recombinant adeno-associated virus vector genomes in muscle. J Virol 2003 ; 77 : 3495–3504. [CrossRef] [PubMed] [Google Scholar]
- Sun X, Lu Y, Bish LT, et al. Molecular analysis of vector genome structures after liver transduction by conventional and self-complementary adeno-associated viral serotype vectors in murine and nonhuman primate models. Hum Gene Ther 2010 ; 21 : 750–761. [CrossRef] [PubMed] [Google Scholar]
- Duan D, Sharma P, Yang J, et al. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J Virol 1998 ; 72 : 8568–8577. [PubMed] [Google Scholar]
- Duan D, Yan Z, Yue Y, Engelhardt JF. Structural analysis of adeno-associated virus transduction circular intermediates. Virology 1999 ; 261 : 8–14. [CrossRef] [PubMed] [Google Scholar]
- Yang J, Zhou W, Zhang Y, et al. Concatamerization of adeno-associated virus circular genomes occurs through intermolecular recombination. J Virol 1999 ; 73 : 9468–9477. [PubMed] [Google Scholar]
- Nakai H, Yant SR, Storm TA, et al. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J Virol 2001 ; 75 : 6969–6976. [CrossRef] [PubMed] [Google Scholar]
- Miller DG, Trobridge GD, Petek LM, et al. Large-scale analysis of adeno-associated virus vector integration sites in normal human cells. J Virol 2005 ; 79 : 11434–11442. [CrossRef] [PubMed] [Google Scholar]
- Nakai H, Montini E, Fuess S, et al. AAV serotype 2 vectors preferentially integrate into active genes in mice. Nat Genet 2003 ; 30 : 297–302. [CrossRef] [Google Scholar]
- Nakai H, Wu X, Fuess S, et al. Large-scale molecular characterization of adeno-associated virus vector integration in mouse liver. J Virol 2005 ; 79 : 3606–3614. [CrossRef] [PubMed] [Google Scholar]
- Bell P, Moscioni AD, McCarter RJ, et al. Analysis of tumors arising in male B6C3F1 mice with and without AAV vector delivery to liver. Mol Ther 2006 ; 14 : 34–44. [CrossRef] [PubMed] [Google Scholar]
- Bell P, Wang L, Lebherz C, et al. No evidence for tumorigenesis of AAV vectors in a large-scale study in mice. Mol Ther 2005 ; 12 : 299–306. [CrossRef] [PubMed] [Google Scholar]
- Donsante A, Miller DG, Li Y, et al. AAV vector integration sites in mouse hepatocellular carcinoma. Science 2007 ; 317 : 477. [CrossRef] [PubMed] [Google Scholar]
- Ranzani M, Cesana D, Bartholomae CC, et al. Lentiviral vector-based insertional mutagenesis identifies genes associated with liver cancer. Nat Methods 2013 ; 10 : 155–161. [CrossRef] [PubMed] [Google Scholar]
- Miller DG, Petek LM, Russell DW. Adeno-associated virus vectors integrate at chromosome breakage sites. Nat Genet 2004 ; 36 : 767–773. [CrossRef] [PubMed] [Google Scholar]
- Kotterman MA, Schaffer DV. Engineering adeno-associated viruses for clinical gene therapy. Nat Rev 2014 ; 15 : 445–451. [CrossRef] [Google Scholar]
- Li H, Malani N, Hamilton SR, et al. Assessing the potential for AAV vector genotoxicity in a murine model. Blood 2011 ; 117 : 3311–3319. [CrossRef] [PubMed] [Google Scholar]
- Tremblay JP. CRISPR, un système qui permet de corriger ou de modifier l’expression de gènes responsables de maladies héréditaires. Med Sci (Paris) 2015 ; 31 : 1014–1022. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des tableaux
Liste des figures
|
Figure 1. Génomes de l’AAV sauvage et recombinant. Le génome de l’AAV de sérotype 2, représenté ici, contient trois promoteurs (p5, p19 et p40) et un signal de polyadénylation (polyA). Il est encadré par deux répétitions terminales, les ITR (pour inverted terminal repeats) qui sont des régions essentielles pour la réplication et l’encapsidation du génome viral. Le génome sauvage code 8 protéines : les protéines régulatrices Rep (gène rep), les protéines structurales VP (gène cap), et la protéine chaperonne d’assemblage AAP. Le génome du vecteur AAV recombinant est obtenu en remplaçant les gènes viraux par une cassette d’expression du transgène qui contient, au minimum, un promoteur, un ADN complémentaire (ADNc) et un signal de polyA. Les ITR sont donc les seules séquences d’origine virale présentes dans le vecteur AAVr. Ces deux types de génomes sont encapsidés, sous la forme d’une molécule d’ADN simple brin, dans une capside virale de 20 nm de diamètre composée des 3 protéines VP formant une particule d’AAV sauvage ou d’AAVr. |
| Dans le texte | |
|
Figure 2. Modèle théorique d’intégration du génome de l’AAV sauvage dans le locus AAVS1. L’ITR (inverted terminal repeats) de l’AAV et le locus AAVS1 du génome cellulaire partagent un site de fixation pour les protéines Rep (ou RBS [Rep binding site]) et un site de clivage par ces mêmes protéines (ou trs, terminal resolution site). Selon le modèle d’intégration, les protéines virales Rep seraient capables de réaliser un pontage entre le génome viral et le génome cellulaire via la reconnaissance du site RBS. La fixation de ces protéines au RBS entraîne un clivage de l’ADN cellulaire et viral au niveau du site trs. Le génome viral s’intégrerait alors via un mécanisme encore mal élucidé impliquant plusieurs facteurs de réparation de l’ADN ainsi que des polymérases cellulaires. Des travaux récents ont montré que l’intégration du génome de l’AAV survenait majoritairement en orientation inverse. Il est important aussi de noter que l’intégration virale peut survenir dans une région chromosomique très large et de façon préférentielle en amont du site RBS. Il en résulte, dans la plupart des cas, une délétion partielle ou complète des ITR ainsi que des délétions et/ou des réarrangements des séquences cellulaires adjacentes. Des sites RBS consensus et des sites trs cryptiques ont été également retrouvés en grand nombre dans d’autres régions chromosomiques, ce qui explique la capacité du virus à cibler différents locus cellulaires (voir Figure 3). |
| Dans le texte | |
|
Figure 3. Distribution du nombre d’événements d’intégration de l’AAV sauvage et recombinant par chromosome. Le nombre d’intégrations du génome AAV sauvage ( A ) ou recombinant ( B ) a été mesuré après infection de fibroblastes primaires humains. Il est exprimé en nombre d’événements d’intégration détectés toutes les 10 mégabases (Mb) d’ADN (© Daniela Hüser et Regine Heilbronn, institut de virologie, université de médecine de la Charité, Berlin, Allemagne). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.