")
")
| Issue |
Med Sci (Paris)
Volume 31, Novembre 2015
Les Cahiers de Myologie
|
|
|---|---|---|
| Page(s) | 20 - 27 | |
| Section | Mise au point | |
| DOI | https://doi.org/10.1051/medsci/201531s306 | |
| Published online | 6 novembre 2015 | |
Myopathie GNE
GNE myopathy
1
Praticien hospitalier, APHP, Hôpital Marin, Hendaye. Centre de Référence GNMH, Chargé de Mission, FILNEMUS, Marseille, France
2
Praticien hospitalier, Institut de MyologieCHU Paris-GH La Pitié Salpêtrière, Centre de Référence de Pathologie Neuromusculaire Paris Est, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
**
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La myopathie GNE est une maladie neuromusculaire rare et de description relativement récente. Elle touche une population majoritairement d’âge adulte et se transmet selon un mode autosomique récessif. Bien que rare et universelle, elle prévaut dans la communauté juive d’origine perse installée en Israël ou aux États-Unis, dans des populations extrême-orientales (Japon et pays avoisinants) et, plus près de chez nous, en Bulgarie. Elle entraîne une faiblesse musculaire prédominant sur les extrémités (myopathie distale), touchant initialement, et de façon prépondérante, les muscles releveurs de pieds. Le terme générique de myopathie GNE fait désormais consensus et recouvre plusieurs entités précédemment décrites : la myopathie respectant le quadriceps, la myopathie à inclusions autosomique récessive (hIBM), la myopathie distale de type Nonaka (ou DMRV pour distal myopathy with rimmed vacuoles). Cette myopathie est due à un dysfonctionnement du gène GNE codant une enzyme bifonctionnelle, l’UDP-N-acétylglucosamine-2-épimerase/N-acétylmannosamine kinase. Celle-ci intervient à deux niveaux dans la voie métabolique aboutissant à la synthèse de l’acide sialique. L’acide sialique, aussi appelé acide N-acétylneuraminique (Neu5Ac ou NANA en abrégé), est un monosaccharide indispensable à d’autres molécules, protéiques ou lipidiques, nécessitant des résidus sucrés à leur surface pour un bon fonctionnement. La myopathie GNE s’accompagne de lésions histologiques (vacuoles bordées) à l’intérieur des fibres musculaires. Celles-ci sont assez typiques dans un contexte clinique évocateur, mais non spécifiques et inconstantes d’un muscle à l’autre. Le diagnostic positif de la myopathie GNE repose sur la clinique, dont l’imagerie musculaire, et sur les études génétiques. Si des essais thérapeutiques prometteurs se développent actuellement pour pallier le défaut métabolique récemment mis au jour, le traitement de cette myopathie reste pour l’instant purement symptomatique.
Abstract
GNE myopathy is a rare neuromuscular disease whose description is fairly recent. It predominantly affects the adult population and is an inherited autosomal recessive disorder. Although universal and ubiquitous, GNE myopathy prevails in the Jewish community of Persian origin, living in Iran, Israel or in the United States. This condition has also been reported in great number in populations of far-East Asia (Japan and neighboring countries) and, closer to France, in Bulgaria. GNE myopathy causes muscle weakness in the extremities (distal myopathy), affecting initially and predominantly foot flexor muscles. The generic term of GNE myopathy is now fully accepted and encompasses two previously described entities: the quadriceps sparing myopathy, (also referred to as the autosomal recessive form of inclusion body myopathy (hIBM) and the Nonaka type distal myopathy (or distal myopathy with rimmed vacuoles DMRV). This myopathy is due to mutations in the GNE gene encoding a bifunctional enzyme, the UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase. This enzyme plays a role at two levels in the metabolic pathway leading to the synthesis of sialic acid. Sialic acid, also known as N-acetylneuraminic acid (Neu5Ac or NANA), is a monosaccharide essential to other protein or lipid molecules requiring sugar residues on their surface in order to function efficiently. GNE myopathy is characterized by histological lesions (rimmed vacuoles) within muscle fibers. They are fairly typical in a suggestive context, but non-specific and inconsistent from one muscle to another. The diagnosis of GNE myopathy is essentially based on clinical clues, including muscle imaging, and is confirmed by genetic studies. If promising therapeutic trials are being developed to compensate for this recently unveiled metabolic defect, the treatment of this myopathy remains purely supportive to date.
© 2015 médecine/sciences – Inserm
Vignette (Photo © Anthony Béhin).
Épidémiologie
La myopathie GNE répond à la définition d’une maladie rare avec une prévalence inférieure à 1 personne atteinte sur 2 000. Toutefois, les chiffres exacts d’incidence ou de prévalence de la maladie restent encore mal connus. Parmi les facteurs explicatifs, on citera le caractère récent de la description de cette nouvelle entité, le défaut de reconnaissance de ce type de myopathie par le corps médical, y compris parmi les spécialistes de pathologie neuromusculaire (les pièges diagnostiques sont nombreux dans la myopathie GNE), et enfin, les imperfections du registre international qui peine à répertorier de manière exhaustive les cas diagnostiqués dans le monde.
Concernant la prévalence, Orphanet avance le chiffre de 0,1 cas pour 100 000 habitants. Cette estimation varie toutefois sensiblement d’une zone géographique à l’autre. En France, on estime que moins d’une centaine de personnes pourraient être concernées par cette myopathie dont une quarantaine a déjà été confirmée en biologie moléculaire (Béhin, 2008). Un quart environ de ces patients vivent dans l’île de la Réunion.
La myopathie GNE est plus fréquente dans au moins cinq pays : au Japon, où elle a été décrite pour la première fois par Nonaka, en Iran, en Israël, aux États-Unis et en Bulgarie.
En Iran, aux États-Unis et en Israël, la grande fréquence des cas rapportés est liée à l’émergence et la dissémination d’une mutation ancestrale du gène GNE (p.Met743Thr) au sein de la minorité juive d’origine perse. De tels patients ont été diagnostiqués en Iran et dans les pays avoisinants (Ouzbékistan, Irak, Afghanistan). Une bonne partie de ces communautés juives d’origine perse a émigré vers Israël, vers la côte ouest des États-Unis, en Californie, et à New York. La même mutation a été retrouvée dans d’autres populations juives, sans lien direct avec l’Iran et installées en Syrie et en Égypte (notamment dans la minorité religieuse karaïte) (Argov 2014).
L’effet fondateur, qui daterait de 2 500 ans, a diffusé plus largement au Moyen-Orient et au Maghreb, en Tunisie notamment (Amouri, 2005), et ce dans des populations non-juives (Haghighi, 2015). Plusieurs familles ont ainsi été diagnostiquées dans des communautés palestiniennes et bédouines du Moyen-Orient établies en Israël, en Arabie Saoudite, au Yémen, et au Koweït. Un article fait état d’un isolat de patients dans une petite ville à l’est de Téhéran (Sangesar) dans une communauté de confession bahaïe (une religion établie au XIXe siècle et dont les liens avec le judaïsme font l’objet de débats) (Khademian, 2013). Au total, plus de 300 patients seraient porteurs de la mutation p.Met743Thr à l’état homozygote, toutes origines confondues.
Le Japon, et dans une moindre mesure le nord de la Chine et la Corée, sont des pays où la myopathie GNE est également rapportée en grand nombre (200 cas recensés pour le seul Japon) (Sim, 2013 ; Zhao, 2015). En revanche, la diversité mutationnelle y est beaucoup plus grande qu’au Moyen-Orient. En Inde (Nalini, 2013), il faut distinguer la partie méridionale et celle plus septentrionale du sous-continent avec deux spectres mutationnels distincts.
En Europe, on retiendra surtout l’existence d’un cluster de patients découvert en Bulgarie par Ivailo Tournev au sein de la communauté Rom (Chamova, 2015). Ces derniers sont porteurs à l’état homozygote d’une mutation décrite également chez des patients indiens originaires du Rajasthan. De façon plus anecdotique, une prévalence élevée de patients a également été rapportée en Irlande du Nord et dans le Nord de l’Angleterre (Chaouch, 2014).
Un registre international de la myopathie GNE a vu récemment le jour dans le cadre d’un partenariat entre une firme pharmaceutique (Ultragenyx, basée à Novato, en Californie) et l’Alliance TREAT-NMD. Les formulaires, traduits en plus de six langues, sont remplis par les patients avec l’aide de leurs médecins. Ce registre vient s’ajouter à un autre registre, plus ancien, mis en place par une association californienne de patients (l’ARM). Au Japon, les patients sont enregistrés dans le registre national REMUDY lequel est interfacé avec le registre international. En France, les patients suivis sont également intégrés dans la base de données générale des centres de référence de maladies rares (CEMARA).
Physiopathologie
La myopathie GNE est due à des anomalies dans le gène GNE. Identifié en 2001 par une équipe israélienne (Eisenberg, 2001), le gène GNE est localisé sur le chromosome 9 et code une protéine ayant une double fonction enzymatique : l’une de type épimérase, l’autre de type kinase, d’où son nom particulièrement complexe : UDP-N-acétylglucosamine-2-épimerase/N-acétylmannosamine kinase (ou GNE en abrégé) (Kayashima, 2002 ; Reinke, 2009 ; Tong, 2009 ; Yardeni, 2011).
La protéine contient, dans sa version longue récemment revue et corrigée, 753 acides aminés (Huizing, 2014) (Figure 2). Elle intervient dans le métabolisme de l’acide sialique, un composé essentiel qui peut être d’origine exogène (apporté par l’alimentation) ou synthétisé par l’organisme lui-même dans le cadre d’une cascade de réactions enzymatiques endogènes (Kurochkina, 2010). L’acide sialique, ou acide 5-N-acétylneuraminique (Neu5Ac), est un monosaccharide déposé, de manière active, à la surface de glycoprotéines ou de glycolipides. Ce processus appelé sialylation se produit dans le réticulum endoplasmique (Krause, 2005). La production d’acide sialique a lieu quant à elle dans le cytosol, sous le contrôle de plusieurs enzymes. Le substrat initial est le N-acétylglucosamine et dérive du glucose. Sous l’action de l’épimérase de la GNE, celui-ci est transformé en N-acétylmannosamine (ManNAC) puis, par la kinase de cette même enzyme, en acide sialique (Neu5Ac). Pour être actif et entrer dans le processus de sialylation, l’acide sialique doit ensuite être couplé à un résidu cytidine monophosphate (CMP-Neu5Ac) sous l’influence d’une synthétase située dans le noyau cellulaire. Le taux de CMP-Neu5Ac exerce un rétrocontrôle sur l’activité du site épimérase de la GNE (Figure 1). La GNE est exprimée dans tous les tissus mais plus particulièrement dans le foie et les muscles. Pour autant, l’activité enzymatique est variable, y compris d’un muscle à l’autre.
 |
Figure 1. Biosynthèse de l’acide sialique. |
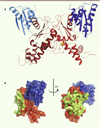 |
Figure 2. Image en cristallographie de la protéine GNE (© Y. Tong et al. PLoS One, october 20, 2009 – © avec l’autorisation de PLoS One). |
La pathogénie de la GNE reste incomplètement connue. En cas de mutation du gène GNE, le taux d’acide sialique est diminué, de 30 à 60 % selon les muscles. Le lien entre cette hyposialylation et le phénotype observé, quasiment exclusivement musculaire, reste obscur. La GNE aurait possiblement d’autres fonctions au sein de l’organisme.
La preuve indirecte de l’implication d’une hyposialylation dans la survenue de la maladie a été apportée par les effets, à certains égards spectaculaires, de la supplémentation en acide sialique chez l’animal.
Dans un premier temps, et faute de disposer d’un mutant naturel, plusieurs modèles animaux transgéniques ont été mis au point (Malicdan, 2007). La souris knock-out pour le gène GNE s’est avérée non viable, preuve du rôle majeur de cette enzyme dans une voie métabolique primordiale. La souris knock-in mise au point par les chercheurs israéliens et portant la mutation humaine moyen-orientale (p.Met743Thr) a développé un phénotype non musculaire, en l’occurrence rénal (glomérulopathie sévère). Une autre souris knock-in contenant la mutation p.Val603Leu, prévalente au Japon, a abouti à un résultat similaire. Seule la souris knock-in incorporant la mutation p.Asp207Val, très présente également en Asie et mise au point par les chercheurs japonais, développe un phénotype très proche de la maladie humaine. Ce modèle Gne Asp207Val, désormais disponible pour les chercheurs et laboratoires du monde entier, a servi et sert de base pour établir la preuve de concept de nombreux essais thérapeutiques.
D’autres modèles animaux sont aussi à l’étude, comme le poisson-zèbre, et font l’objet de travaux plus fondamentaux comme la recherche, non complétement aboutie à ce jour, des partenaires potentiels de la GNE.
Aspects cliniques
La myopathie GNE est une maladie musculaire relativement rare, vraisemblablement sous-diagnostiquée et/ou diagnostiquée tardivement. Et cela malgré un tableau clinique et histopathologique assez évocateur.
Les premiers signes apparaissent généralement à la troisième décennie, même si des débuts plus précoces (à l’adolescence) ont pu être rapportés. La présentation la plus classique est celle d’un déficit progressif des releveurs du pied, parfois unilatéral au début. L’extension aux muscles de la loge postérieure est fréquente et généralement rapide. Il est beaucoup plus rare que les membres supérieurs soient atteints en premier. La progression du processus dégénératif vers le haut aboutit plus ou moins rapidement à une atteinte proximo-distale associant steppage et dandinement caractéristiques. Ceci contraste, même à un stade très avancé de la maladie, avec le respect du volume et de la force du muscle quadricipital (Figure 3). Cette formule sémiologique inhabituelle a été à la base des premières descriptions cliniques (Z. Argov rapporte régulièrement le cas de ses propres patients d’âge mûr, pourtant en fauteuil roulant, capables de faire tenir leurs petits-enfants sur leur jambe tendue…). Une atteinte du quadriceps, d’ailleurs variable en intensité, n’est décrite que dans 5 % des cas. Quelques rares patients se présentent comme des myopathies dites des ceintures, dans lesquelles l’atteinte distale est parfois absente ou plus subtile à détecter cliniquement (Park, 2012). De tels cas n’ont pas été observés en France jusqu’à présent. L’atteinte des membres supérieurs est plus volontiers de topographie distale (déficit des mains, et notamment des fléchisseurs de doigts) et plus tardivement scapulaire (défaut d’abduction, décollement des omoplates plus rarement). Le déficit peut parfois être asymétrique.
 |
Figure 3. À l’examen clinique, la conjonction d’un déficit des muscles releveurs de pied (foot drop) et d’une parfaite conservation du muscle quadriceps doit faire évoquer une myopathie GNE. |
L’atteinte cardiaque ne fait pas partie du tableau classique de la myopathie GNE. Elle a toutefois été rapportée à plusieurs reprises, notamment dans la cohorte de patients décrits en Bulgarie, sans que l’on puisse éliminer formellement une association fortuite. Un suivi cardiologique régulier des patients fait néanmoins partie des recommandations d’experts. Il en est de même pour l’atteinte respiratoire que l’on observe exceptionnellement, essentiellement chez des personnes en fauteuil roulant (Mori-Yoshimura, 2013). Dans certains cas exceptionnels, et très tardivement dans l’évolution, il a pu être noté un petit déficit au niveau des muscles du visage et de ceux du cou. Quant aux fonctions cognitives, elles sont toujours conservées, sauf trouble associé.
Histologie
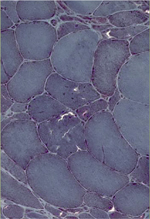
Trois lésions histologiques caractérisent la myopathie GNE. Les vacuoles bordées, l’accumulation de dépôts protéiques et des inclusions filamentaires. Les vacuoles bordées apparaissent en hématine-éosine mais surtout au trichrome modifié de Gomori. Improprement appelées « vacuoles » et donnant l’impression d’être vides en microscopie optique, elles sont en fait le reflet d’un processus d’autophagie pathologique avec accumulation de protéines. Les protéines accumulées y sont pour l’essentiel ubiquitinées et témoignent d’un phénomène d’agréphagie défectueux. Ces dépôts protéiques comprennent entre autres l’alpha-synucléine, la protéine β-amyloïde, et la protéine tau (Figure 5). Les vacuoles bordées ne sont pas spécifiques et peuvent être observées dans de nombreux processus pathologiques (autres myopathies, y compris dans les myosites à inclusions) ou physiologiques (vieillissement). Elles ne prennent vraiment une valeur diagnostique que dans le contexte clinique particulier de la myopathie GNE, à savoir celui d’une myopathie distale avec respect du quadriceps. Prédominant dans les muscles les plus atteints cliniquement, elles n’ont pas de réelle valeur pronostique. En d’autres termes, il n’existe pas de corrélation entre le nombre de fibres musculaires avec vacuoles bordées et la sévérité ou l’évolutivité de la myopathie.
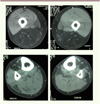 |
Figure 4. En imagerie (ici, un scanner musculaire), l’association d’une atteinte des muscles de la loge postérieure de cuisse et de la loge antérieure de jambe est très évocatrice de myopathie GNE. |
 |
Figure 5. Images de biopsie musculaire (colorations en trichrome Gomori modifié, et en hématoxylline-éosine). Les vacuoles bordées à l’intérieur de fibres musculaires atrophiques sont, à défaut d’être pathognomoniques, évocatrices de myopathie GNE. |
Dans les vacuoles situées dans le cytoplasme et/ou à l’intérieur même des noyaux cellulaires, des inclusions de nature filamentaire sont souvent observées. En microscopie électronique, elles ont généralement 12 à 18 microns de diamètre et côtoient les agrégats protéiques. Les phénomènes inflammatoires sont très exceptionnellement notés à la périphérie des fibres musculaires, de même que les phénomènes de nécrose-dégénération (ces derniers pouvant, le cas échéant, faire discuter un processus dystrophique). Une inflammation importante avec réexpression des protéines de classe I s’observe très exceptionnellement et fait discuter des formes frontières avec les formes sporadiques, d’origine auto-immune, de myosites à inclusions.
Malheureusement, et trop souvent, la biopsie musculaire peut être prise en défaut, surtout si elle porte sur un muscle non atteint, comme le quadriceps, ou tardivement atteint comme le deltoïde. L’idéal serait de prélever un muscle fréquemment atteint comme le jambier antérieur, mais pas trop tardivement tout de même, ou bien alors un muscle gastrocnémien. L’imagerie est, dans ce contexte, particulièrement utile pour guider le geste biopsique.
Les autres examens
L’imagerie musculaire constitue aujourd’hui une aide précieuse au diagnostic de myopathie GNE (Tasca, 2012). Qu’il s’agisse d’images produites en tomodensitométrie ou en IRM, la grande sélectivité de l’atteinte musculaire apparaît au premier plan, au moins aux stades de début. En IRM, les muscles cibles (muscles de la jambe, notamment tibial antérieur, muscles de la loge postérieure de cuisse) se rehaussent d’abord en séquence STIR-T2, puis sont remplacés, au fil des mois ou des années, par du tissu adipeux apparaissant en hyper-signal T1 (Figure 4). On note que l’atteinte est ascendante et touche petit à petit les muscles pelviens. Dans tous les cas, le remarquable respect du quadriceps doit attirer l’attention et faire évoquer une myopathie GNE. A l’ère des essais cliniques, l’IRM peut également servir pour suivre l’évolution de la maladie (histoire naturelle) et mesurer l’effet des traitements.
Les autres examens n’ont qu’une valeur d’orientation : les taux de CPK sont inconstamment élevés (et presque toujours inférieurs à 1 000 unités par litre) et L’EMG est myogène, parfois mixte, notamment à un stade précoce. Quant aux dosages sanguins ou urinaires de l’acide sialique libre, ils sont inconstamment diminués et relèvent du domaine de la recherche.
Aspects génétiques
La myopathie GNE est une maladie génétique transmise selon le mode autosomique récessif. On dénombre à ce jour plus de 160 mutations différentes du gène GNE, dont quatre au moins ont un caractère fondateur dans les pays ou communautés où elles ont été identifiées.
Ces anomalies sont dans leur immense majorité (plus de 95 % des cas) des mutations faux-sens, homozygotes ou hétérozygotes composées. Il n’a jamais été rapporté de mutations stop à l’état homozygote, laissant penser que celles-ci sont létales à l’état embryonnaire. Les mutations peuvent concerner indifféremment le domaine kinase ou le domaine épimérase de l’enzyme.
La liste des transcrits codés par le gène GNE est longue et non exhaustive (8 transcrits rapportés à ce jour dans les bases de données). Ce qui a abouti récemment à une refonte de la nomenclature en matière mutationnelle, un exon supplémentaire du gène GNE ayant été mis à jour. En conséquence, et à titre d’exemple, la mutation moyen-orientale p.Met712Thr est désormais référencée M743G. L’identification des mutations se fait par séquençage direct du gène. De plus en plus, le gène GNE est incorporé dans les panels de gènes ciblés analysés par séquençage à haut débit pour le diagnostic de pathologies musculaires de topographie distale. Plus exceptionnel, le séquençage de l’exome-entier peut également amener au diagnostic moléculaire de myopathie GNE.
En l’absence d’autre biomarqueur fiable, la découverte de mutations pathogènes du gène GNE reste le critère de certitude en matière de diagnostic. Il arrive, très rarement il est vrai, qu’une seule mutation sur les deux soit identifiée. Des anomalies plus subtiles du gène, y compris dans les zones introniques, doivent alors être recherchées. En France, le laboratoire de génétique du CHU de la Timone à Marseille est devenu la référence en matière de diagnostic moléculaire.
Si la pénétrance de la myopathie GNE est quasi-complète, il existe quelques très rares exceptions. Z. Argov rapporte le cas exceptionnel d’un patient israélien qui, bien qu’homozygote pour la mutation moyen-orientale, est, à 78 ans passés, toujours asymptomatique. Quant aux personnes hétérozygotes pour une seule mutation délétère, il n’y a pas de symptomatologie associée (à la différence du modèle animal correspondant chez qui la sialylation est légèrement diminuée).
Dans certains isolats génétiques où la prévalence de la maladie est très significative, le dépistage des hétérozygotes pourrait être éthiquement envisageable, notamment dans le cadre d’un conseil génétique prémarital.
Enfin, des phénomènes de transmission pseudo-dominante de la myopathie GNE ont été observés dans des communautés très endogames, notamment au Moyen-Orient, et sont souvent à la source d’une importante errance diagnostique.
Un peu d’histoire

Zohar Argov (Jérusalem, Israël) et Ikuya Nonaka (Tokyo, Japon) désormais unis pour une même et unique cause, la myopathie GNE.
L’histoire de la myopathie GNE est indissociable de celle de deux myologues de renom : Ikuya Nonaka et Zohar Argov. C’est en 1981 que I. Nonaka, un des meilleurs spécialistes japonais de pathologie musculaire (appartenant au National Center of Neurology and Psychiatry – NCNP - de Tokyo), rapporte les trois premiers cas myopathie GNE dans deux familles distinctes. Il en déduit qu’il s’agit d’une entité nouvelle, du fait d’une topographie singulière (atteinte des muscles jambiers antérieurs) et d’une présence d’anomalies histologiques insolites, bien que non spécifiques, appelées vacuoles bordées. Il la rapporte à l’époque comme une « myopathie distale avec vacuoles bordées de transmission autosomique récessive » (DMRV pour distal myopathy with rimmed vacuoles). Il tient à ce qu’on la distingue d’une autre myopathie distale, également décrite quelques années auparavant par un autre japonais, Kazuo Miyoshi.
Quelques années plus tard, en 1984, une étude portant sur quatre familles d’origine juive perse présentant une myopathie autosomique récessive à vacuoles bordées de transmission autosomique récessive est publiée par Zohar Argov et son équipe de l’hôpital Hadassah de Jérusalem. Les auteurs dont les premières observations remontent à quelques années auparavant, estiment qu’il s’agit d’un nouveau type de myopathie. Ils revendiquent le terme d’« hereditary Inclusion Body Myopathy » (h-IBM), soit en français, la forme héréditaire de myopathie à inclusions. Ils soulignent une caractéristique très frappante à savoir le respect, même à un stade très avancé de la maladie, du muscle de la loge antérieure de cuisse (le muscle quadriceps).
Pendant plusieurs années, Japonais et Israéliens s’opposeront sur un possible apparentement entre ces deux entités (DRMV et h-IBM). En parallèle, des chercheurs californiens, V. Askanas et A. Engel, travaillaient sur une forme très proche de l’h-IBM mais dans laquelle l’origine était clairement auto-immune. Cette forme portait l’acronyme de s-IBM (pour sporadic Inclusion Body Myositis) et présentait des similitudes avec les lésions cérébrales observées dans la maladie d’Alzheimer (agrégats protéiques, inclusions filamentaires, etc.). Une théorie est même échafaudée à l’époque selon laquelle la s-IBM constituerait une sorte de maladie d’Alzheimer du muscle, l’h-IBM en représentant la variante familiale (à l’instar de ce que l’on observe aussi dans les formes familiales de maladie d’Alzheimer).
Il faudra finalement attendre 2001 et la découverte des mutations dans le gène GNE par Stella Mitrani-Rozenbaum, la collaboratrice directe de Zohar Argov formée à l’Institut Pasteur de Paris, pour que les phénotypes observés au Japon et en Israël soient bien réunis au sein d’une seule et même entité clinique, la myopathie GNE (cette appellation faisant désormais consensus depuis 2013).
Diagnostic différentiel
En regard de l’expérience française mais aussi internationale, rares sont les myopathies qui ont donné lieu à autant d’erreurs diagnostiques que la myopathie GNE. Ceci aboutit, encore parfois de nos jours, à une errance diagnostique pouvant atteindre 30 ans chez certains patients. Beaucoup de facteurs concourent à cette constatation : le caractère parfois discret et/ou ancien de l’atteinte distale, l’absence d’antécédents familiaux, des arguments ténus en faveur d’une myopathie plutôt que d’une neuropathie, des données électrophysiologiques contradictoires, l’absence de vacuoles bordées à l’histologie, etc.
L’affirmation du caractère distal du processus myopathique est une première étape importante dans la démarche diagnostique. Ceci permet de discuter, dans les cas atypiques, d’autres étiologies. La question se pose rarement concernant la myopathie distale de type Miyoshi (déficit en dysferline) ou sa variante, l’anoctaminopathie (déficit en anoctamine 5) : si la transmission est la même (autosomique récessive), les taux de CPK sont nettement plus élevés et la topographie de l’atteinte beaucoup plus marquée, sauf exception, au niveau de la loge postérieure de jambe. Les lésions histologiques sont aussi beaucoup plus « dystrophiques » que dans la myopathie GNE. Les autres myopathies distales à CPK normales ou peu élevées sont à considérer le cas échéant : il s’agit notamment des formes récessives de titinopathies, ou plus rarement des myopathies normalement transmises selon un mode autosomique dominant (myopathie de Welander, myopathies myofibrillaires).
À un stade plus avancé, quand l’atteinte proximale des membres inférieurs devient prépondérante, on s’interrogera sur un possible diagnostic de myopathie des ceintures. L’atteinte préférentielle des loges postérieures de cuisse se discute notamment avec une calpainopathie primitive
Une observation fait également état d’une confusion possible avec une amyotrophie spinale de type 3.
En cas de transmission pseudo-dominante de myopathie GNE, certaines myopathies des ceintures, la myopathie facio-scapulo-humérale (FSH), les myopathies myofibrillaires et certains syndromes scapulo-péroniers pourront être évoqués.
Reste le cas, au demeurant le plus fréquent, de la confusion entre une myopathie distale et une forme spinale de maladie de Charcot-Marie-Tooth de transmission autosomique récessive. Là aussi, il faut savoir évoquer une myopathie GNE au moindre doute.
Chez les personnes chez qui la maladie se déclare à un stade beaucoup plus tardif, et notamment si l’histologie révèle la présence d’infiltrats inflammatoires, certains auteurs ont pu discuter le diagnostic de myosite en général, et de myosite à inclusions en particulier.
La myopathie autosomique dominante VCP associant une formule histologique de type IBM, une maladie de Paget et une démence fronto-temporale peut faire également partie du diagnostic différentiel si les deux dernières composantes manquent au tableau et s’il s’agit d’un cas sporadique.
Dans tous ces cas difficiles, l’imagerie musculaire est très souvent d’un apport majeur, sauf à un stade très avancé de la maladie. Le recours au séquençage du gène GNE en est un autre, celui-ci étant relativement simple, fiable, et peu coûteux.
Recherche et essais thérapeutiques
L’histoire naturelle de la myopathie GNE reste à écrire. Une étude pilotée par le NIH américain a été lancée en 2011 et vise à mesurer dans le temps les paramètres cliniques et biologiques les plus pertinents en vue des essais thérapeutiques. Les chercheurs n’ont pas identifié à ce jour de biomarqueurs fiables et corrélant bien avec l’évolution de la maladie. Une autre étude, similaire dans ses objectifs, est en cours au niveau international, avec le soutien du laboratoire Ultragenyx.
Même si la physiopathologie de la myopathie GNE est imparfaitement comprise, le rationnel de la grande majorité des essais thérapeutiques repose actuellement sur la restauration de la sialylation à l’échelle cellulaire (Nishino, 2015 ; Yonekawa, 2014).
Pour valider cette hypothèse, et outre les études précliniques encourageantes chez les souris transgéniques (Malicdan, 2009 ; Malicdan, 2010), un premier essai a consisté à administrer des cures intraveineuses d’immunoglobulines, celles-ci étant connues pour leur concentration élevée en acide sialique. L’étude a été réalisée en ouvert au NIH en 2005 chez quatre individus avec des résultats cliniques intéressants, mais sans modification des lésions histologiques ni du taux de glycoprotéines étudiées (Sparks, 2007).
Un essai plus récent au Japon a montré que l’administration orale d’acide sialique (800 mg trois fois par jour) était bien tolérée.
En 2012, un essai de phase II a été promu et réalisé par Ultragenyx. Ce laboratoire a développé une forme retard d’acide sialique permettant de contrecarrer la tendance physiologique de ce dernier à être excrété par le rein quand il est administré en excès. Réalisé en double aveugle sur une période de 6 mois, il a donné des résultats suffisamment probants pour qu’une phase III soit désormais en cours. Les doses quotidiennes utilisées étaient de 3 g et 6 g d’acide sialique retard.
Une autre approche consiste à utiliser le précurseur de l’acide sialique, le N-acétylmannosamine (ou ManNAc), pour tenter de forcer la machinerie enzymatique. L’effet s’est avéré positif avec une bonne tolérance clinique. Un premier essai de phase I avait commencé au Japon mais avait dû être interrompu faute de production du produit (le laboratoire ayant souffert du tremblement de terre de la région du Kasai).
D’autres approches thérapeutiques existent et reposent pour l’essentiel sur l’apport du gène GNE sain. Un lipoplex, construction de thérapie génique associant un gène-médicament (ici le gène GNE sain sous le contrôle d’un promoteur CMV) et un vecteur de type liposome, a été administré pendant 13 mois à une malade âgée de 41 ans, d’abord par voie intramusculaire puis par voie intraveineuse, le tout avec une bonne tolérance clinique et biologique (Nemunaitis, 2011). L’état, déjà avancé, de la patiente, n’a pas permis de tirer de conclusion utile au niveau clinique.
D’autres études sont en cours chez l’animal, principalement dans l’équipe israélienne de Z. Argov où existe un programme de thérapie génique GNE chez l’animal (Mitrani-Rosenbaum, 2012).
Comment est organisée la recherche dans la myopathie par déficit en GNE ?
La recherche dans la myopathie GNE est particulièrement active dans les pays où la fréquence de la maladie est, pour des raisons historiques, ethniques ou religieuses, plus élevée qu’à l’habitude. C’est en Israël, au Japon et aux Etats-Unis, et plus particulièrement en Californie, que plusieurs équipes de laboratoire sont dédiés spécifiquement à ce type de myopathie. Les Européens sont un peu plus en retrait au niveau de la recherche fondamentale mais prennent toutes leur place dans les essais multicentriques qui se mettent en place à l’échelle internationale. Au niveau français, des chercheurs de l’Institut de Myologie (Anthony Béhin, Bruno Eymard) et du laboratoire de génétique médicale de la Timone à Marseille (Martin Krahn, Nicolas Lévy) s’intéressent de près au sujet depuis plusieurs années.
Un consortium international s’est également créé, réunissant, à intervalles réguliers, chercheurs fondamentalistes, généticiens, cliniciens et industriels du médicament. La dernière réunion a eu lieu en octobre 2014, en marge du congrès de la World Muscle Society de Berlin.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Amouri R, Driss A, Murayama K, et al. Allelic heterogeneity of GNE gene mutation in two Tunisian families with autosomal recessive inclusion body myopathy. Neuromuscul Disord 2005 ; 15 : 361–363. [CrossRef] [PubMed] [Google Scholar]
- Argov Z. GNE myopathy: a personal trip from bedside observation to therapeutic trials. Acta Myol 2014 ; 33 : 107–110. [PubMed] [Google Scholar]
- Béhin A, Dubourg O, Laforêt P, et al. Distal myopathy due to mutations of GNE gene: clinical spectrum and diagnosis. Rev Neurol (Paris) 2008 ; 164 : 434–443. [CrossRef] [PubMed] [Google Scholar]
- Chamova T, Guergueltcheva V, Gospodinova M, et al. GNE myopathy in Roma patients homozygous for the p. I618T founder mutation. Neuromuscul Disord 2015 ; 25 : 713–718. [CrossRef] [PubMed] [Google Scholar]
- Chaouch A, Brennan KM, Hudson J, et al. Two recurrent mutations are associated with GNE myopathy in the North of Britain. J Neurol Neurosurg Psychiatry 2014 ; 85 : 1359–1365. [CrossRef] [PubMed] [Google Scholar]
- Eisenberg I, Avidan N, Potikha T, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet 2001 ; 29 : 83–87. [CrossRef] [PubMed] [Google Scholar]
- Haghighi A, Nafissi S, Qurashi A, et al. Genetics of GNE myopathy in the non-Jewish Persian population. Eur J Hum Genet 2015 May 13. doi: 10.1038/ejhg.2015.78 [Google Scholar]
- Huizing M, Carrillo-Carrasco N, Malicdan MC, et al. GNE myopathy: new name and new mutation nomenclature. Neuromuscul Disord 2014 ; 24 : 387–389. [CrossRef] [PubMed] [Google Scholar]
- Kayashima T, Matsuo H, Satoh A, et al. Nonaka myopathy is caused by mutations in the UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase gene (GNE). J Hum Genet 2002 ; 47 : 77–79. [CrossRef] [PubMed] [Google Scholar]
- Khademian H, Mehravar E, Urtizberea J, et al. Prevalence of GNE p. M712T and hereditary inclusion body myopathy (HIBM) in Sangesar population of Northern Iran. Clin Genet 2013 ; 84 : 589–592. [CrossRef] [PubMed] [Google Scholar]
- Krause S, Hinderlich S, Amsili S, et al. Localization of UDP-GlcNAc 2-epimerase/ManAckinase (GNE) in the Golgi complex and the nucleus of mammalian cells. Exp Cell Res 2005 ; 304 : 365–379. [CrossRef] [PubMed] [Google Scholar]
- Kurochkina N, Yardeni T, Huizing M. Molecular modeling of the bifunctional enzyme UDP-GlcNAc 2-epimerase/ManNAc kinase and predictions of structural effects of mutations associated with HIBM and sialuria. Glycobiology 2010 ; 20 : 322–337. [CrossRef] [PubMed] [Google Scholar]
- Malicdan MC, Noguchi S, Nonaka I, et al. A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum Mol Genet 2007 ; 16 : 2669–2682. [CrossRef] [PubMed] [Google Scholar]
- Malicdan MC, Noguchi S, Nishino I. A preclinical trial of sialic acid metabolites on distal myopathy with rimmed vacuoles/hereditary inclusion body myopathy, a sugar-deficient myopathy: a review. Ther Adv Neurol Disord 2010 ; 3 : 127–135. [CrossRef] [PubMed] [Google Scholar]
- Mori-Yoshimura M, Oya Y, Hayashi YK, et al. Respiratory dysfunction in patients severely affected by GNE myopathy (distal myopathy with rimmed vacuoles). Neuromuscul Disord 2013 ; 23 : 84–88. [CrossRef] [PubMed] [Google Scholar]
- Mitrani-Rosenbaum S, Yakovlev L, Becker Cohen M, et al. Sustained expression and safety of human GNE in normal mice after gene transfer based on AAV8 systemic delivery. Neuromuscul Disord 2012 ; 22 : 1015–1024. [CrossRef] [PubMed] [Google Scholar]
- Malicdan MC, Noguchi S, Hayashi YK, et al. Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV-hIBM mouse model. Nat Med 2009 ; 15 : 690–695. [CrossRef] [PubMed] [Google Scholar]
- Nalini A, Gayathri N, Nishino I, et al. GNE myopathy in India. Neurol India 2013 ; 61 : 371–374. [CrossRef] [PubMed] [Google Scholar]
- Nishino I, Carrillo-Carrasco N, Argov Z. GNE myopathy: current update and future therapy. J Neurol Neurosurg Psychiatry 2015 ; 86 : 385–392. [CrossRef] [PubMed] [Google Scholar]
- Nemunaitis G, Jay CM, Maples PB, et al. Hereditary inclusion body myopathy: single patient response to intravenous dosing of GNE gene lipoplex. Hum Gene Ther 2011 ; 22 : 1331–1341. [CrossRef] [PubMed] [Google Scholar]
- Park YE, Kim HS, Choi ES, et al. Limb-girdle phenotype is frequent in patients with myopathy associated with GNE mutations. J Neurol Sci 2012 ; 321 : 77–81. [CrossRef] [PubMed] [Google Scholar]
- Reinke SO, Lehmer G, Hinderlich S, et al. Regulation and pathophysiological implications of UDP-GlcNAc 2-epimerase/ManNAc kinase (GNE) as the key enzyme of sialic acid biosynthesis. Biol Chem 2009 ; 390 : 591–599. [CrossRef] [PubMed] [Google Scholar]
- Sim JE, Park HJ, Shin HY, et al. Clinical characteristics and molecular genetic analysis of Korean patients with GNE myopathy. Yonsei Med J 2013 ; 54 : 578–582. [CrossRef] [PubMed] [Google Scholar]
- Sparks S, Rakocevic G, Joe G, et al. Intravenous immune globulin in hereditary inclusion body myopathy: a pilot study. BMC Neurol 2007 ; 7 : 3. [PubMed] [Google Scholar]
- Tasca G, Ricci E, Monforte M, et al. Muscle imaging findings in GNE myopathy. J Neurol 2012 ; 259 : 1358–1365. [CrossRef] [PubMed] [Google Scholar]
- Tong Y, Tempel W, Nedyalkova L, et al. Crystal structure of the N-acetylmannosamine kinase domain of GNE. PLoS One 2009 ; 4 : e7165. [CrossRef] [PubMed] [Google Scholar]
- Yardeni T, Choekyi T, Jacobs K, et al. Identification, tissue distribution, and molecular modeling of novel human isoforms of the key enzyme in sialic acid synthesis, UDP-GlcNAc 2-epimerase/ManNAc kinase. Biochemistry 2011 ; 50 : 8914–8925. [CrossRef] [PubMed] [Google Scholar]
- Yonekawa T, Malicdan MC, Cho A, et al. Sialyllactose ameliorates myopathic phenotypes in symptomatic GNE myopathy model mice. Brain 2014 ; 137 : 2670–2679. [CrossRef] [PubMed] [Google Scholar]
- Zhao J, Wang Z, Hong D, et al. Mutational spectrum and clinical features in 35 unrelated mainland Chinese patients with GNE myopathy. J Neurol Sci 2015 ; 354 : 21–26. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Biosynthèse de l’acide sialique. |
| Dans le texte | |
|
Figure 2. Image en cristallographie de la protéine GNE (© Y. Tong et al. PLoS One, october 20, 2009 – © avec l’autorisation de PLoS One). |
| Dans le texte | |
|
Figure 3. À l’examen clinique, la conjonction d’un déficit des muscles releveurs de pied (foot drop) et d’une parfaite conservation du muscle quadriceps doit faire évoquer une myopathie GNE. |
| Dans le texte | |
|
Figure 4. En imagerie (ici, un scanner musculaire), l’association d’une atteinte des muscles de la loge postérieure de cuisse et de la loge antérieure de jambe est très évocatrice de myopathie GNE. |
| Dans le texte | |
|
Figure 5. Images de biopsie musculaire (colorations en trichrome Gomori modifié, et en hématoxylline-éosine). Les vacuoles bordées à l’intérieur de fibres musculaires atrophiques sont, à défaut d’être pathognomoniques, évocatrices de myopathie GNE. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.