")
")
| Issue |
Med Sci (Paris)
Volume 31, Number 10, Octobre 2015
|
|
|---|---|---|
| Page(s) | 912 - 919 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20153110018 | |
| Published online | 19 octobre 2015 | |
Le poisson zèbre
Un modèle d’étude des dystrophies musculaires congénitales
Potential of the zebrafish model to study congenital muscular dystrophies
Division of biological sciences, university of California, San Diego, 9500 Gilman Drive, La Jolla, CA
92093-0380, La Jolla, États-Unis
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Dans le but de mieux comprendre la complexité des dystrophies musculaires congénitales (DMC) et de développer des stratégies pour les traiter, il est important d’établir de nouveaux modèles de ces maladies. En raison de ses nombreuses caractéristiques, le poisson zèbre est récemment devenu un modèle de choix dans l’étude des DMC. La modélisation de certaines de ces dystrophies dans ce modèle vertébré permet en effet d’obtenir un phénotype plus proche de la maladie humaine que le modèle murin. Ces dernières années, grâce à des moyens techniques innovants, il est devenu possible de cribler rapidement et à grande échelle les pathologies musculaires dans ce modèle. Par ailleurs, les nouvelles techniques de modification du génome du poisson ouvrent la voie vers la création de nouveaux modèles pour analyser les DMC. Cette revue discute des qualités majeures du poisson zèbre pour l’étude des DMC et présente les principaux modèles de dystrophies obtenus dans ce modèle.
Abstract
In order to better understand the complexity of congenital muscular dystrophies (CMD) and develop new strategies to cure them, it is important to establish new disease models. Due to its numerous helpful attributes, the zebrafish has recently become a very powerful animal model for the study of CMD. For some CMD, this vertebrate model is phenotypically closer to human pathology than the murine model. Over the last few years, researchers have developed innovative techniques to screen rapidly and on a large scale for muscle defects in zebrafish. Furthermore, new genome editing techniques in zebrafish make possible the identification of new disease models. In this review, the major attributes of zebrafish for CMD studies are discussed and the principal models of CMD in zebrafish are highlighted.
© 2015 médecine/sciences – Inserm
Les dystrophies musculaires congénitales (DMC) sont un groupe hétérogène de maladies neuromusculaires. L’incidence des DMC, toutes formes confondues, est estimée à 1 naissance sur 21 500 [1]. Elles affectent spécifiquement les muscles squelettiques et se caractérisent par une dégénérescence et un affaiblissement progressif de ces muscles [2].
Les découvertes concernant les DMC se sont largement développées cette dernière décennie. Elles ont permis l’identification de nouveaux gènes impliqués dans ces maladies dégénératives, ouvrant la voie vers de nouvelles perspectives thérapeutiques. Cependant, malgré des progrès constants permettant d’améliorer la qualité de vie des patients, aucun traitement curatif n’a pu, pour le moment, être développé avec succès. Il est donc important de mieux comprendre les bases moléculaires de ces pathologies et de développer des modèles animaux mimant la pathogenèse de la maladie chez l’homme, afin de caractériser de nouveaux gènes d’intérêt, de comprendre l’évolution de la maladie et de développer de nouvelles thérapies. Le modèle vertébré du poisson zèbre (Danio rerio) a montré son intérêt dans ces avancées [51]1.
Dans cette revue, nous décrirons les différents aspects des DMC. Nous énumérerons ensuite les moyens mis en œuvre pour analyser ces pathologies dans le modèle de choix que représente le poisson zèbre. Nous discuterons enfin de la pertinence de certaines études réalisées avec ce modèle, offrant un intérêt particulier pour la compréhension des DMC.
Les dystrophies musculaires congénitales
L’hétérogénéité des DMC est illustrée par la variabilité de leurs signes cliniques, mais également par l’âge d’apparition des premiers symptômes. L’étiologie de ces maladies est diverse et complexe. Des mutations différentes d’un même gène peuvent entraîner des manifestations cliniques très variées, et cela au sein d’une même famille ; à l’inverse, des symptômes similaires peuvent résulter d’altérations génomiques distinctes [3]. Outre la classification clinique, les DMC sont aussi classées selon les déficits moléculaires qui les caractérisent (dystroglycanopathies, laminopathies, etc.). À ce jour, les défauts génétiques de plus de vingt-cinq types de DMC ont été identifiés [4].
Au niveau du myosepte vertical du poisson zèbre (voir Glossaire), qui est l’analogue anatomique du tendon des mammifères, se situent la membrane basale, constituée de laminine et de collagène, et les complexes d’adhésion, formés de dystroglycanes et d’intégrines, sur lesquels sont ancrées les fibres musculaires (Figures 1 et 2). Les sous-types de DMC ont pour origine des mutations touchant des gènes codant les protéines se liant à la lame basale, comme l’intégrine α7, ou ceux codant des protéines du complexe de glycoprotéines associé à la dystrophine (Figure 2). D’autres mutations touchent des protéines constituant la lame basale, comme le collagène VI ou la laminine α2 (mérosine).
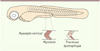 |
Figure 1. Schéma d’un embryon de poisson zèbre deux jours après fécondation. . Lorsqu’un processus dystrophique apparaît, la stabilité structurale de l’ancrage des myofibres est affectée et les fibres se détachent du myosepte vertical. |
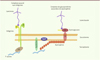 |
Figure 2. Schéma simplifié des complexes d’adhésion de la jonction myotendineuse . Les complexes d’adh販on permettent de relier le cytosquelette d’actine des myofibres à la lame basale. Ces différents complexes se compensent l’un l’autre. CaV3 : caveolin-3 ; ILK : integrin-linked kinase. |
Un groupe particulier de DMC, les dystroglycanopathies, a récemment été mis en évidence. Il regroupe des défauts touchant des protéines qui sont impliquées dans la glycosylation de l’α-dystroglycane [5]. D’autres DMC impliquent un déficit en sélénoprotéine N, codée par le gène sepn1, ou un déficit en lamine A/C, codée par le gène lmna.
Le poisson zèbre, un modèle d’étude des dystrophies musculaires congénitales
Depuis le début des années 1990, le poisson zèbre est devenu un modèle incroyablement utile dans l’étude des DMC. Un des avantages de ce modèle est qu’il est possible, lors des croisements, d’engendrer un grand nombre d’individus ex utero. En effet, une ponte produit de 50 à 200 embryons. D’autres avantages du poisson zèbre incluent son développement rapide, sa facilité de manipulation et sa transparence. Ces propriétés uniques en font un puissant modèle vertébré pour le traçage génétique et les analyses in vivo à l’échelle de la cellule [6].
La possibilité d’utiliser des morpholinos (voir Glossaire) pour manipuler génétiquement le danio représente également un outil important pour identifier rapidement des gènes potentiellement impliqués dans la pathologie. L’embryon du poisson ayant la capacité d’absorber des médicaments, les criblages à grande échelle de molécules pharmacologiques, permettant de définir des gènes cibles importants, sont possibles. Outre ces caractéristiques générales, le poisson zèbre présente d’autres potentialités qui sont essentielles pour l’étude des DMC humaines. Ainsi, les modèles génétiques réalisés chez ce poisson présentent des similitudes importantes avec la pathogenèse des dystrophies musculaires de l’homme [7]. Les phénotypes observés sont plus proches en effet de ceux observés chez l’homme, notamment la sévérité des signes cliniques, que dans certains modèles murins [8, 9]. Ainsi, contrairement au phénotype musculaire du modèle murin mdx, invalidé pour le gène de la dystrophine, qui est modéré et ne compromet pas la survie, le phénotype moteur résultant de l’inactivation du même gène chez le poisson est sévère à quatre jours de développement, et le poisson meurt à deux semaines de développement [10, 11]. Le corps du poisson est composé en majorité de muscles. Les fibres musculaires lentes et rapides s’étendent le long de l’axe antéropostérieur des somites (voir Glossaire) [12]. La séparation entre deux somites, où les fibres musculaires s’attachent, est appelée myosepte vertical (Figure 1). Beaucoup de caractéristiques moléculaires et histologiques, notamment ultrastructurales, sont partagées par les muscles squelettiques humains et ceux du poisson zèbre. Par exemple, on retrouve chez les deux espèces le complexe moléculaire d’excitation-contraction et l’appareil contractile, ainsi que les composants du complexe de glycoprotéines associées à la dystrophine. L’existence de ces deux complexes chez le poisson est donc essentielle pour étudier la pathogenèse des myopathies [13–15]. Les gènes importants pour le développement des muscles et leur intégrité ultérieure sont également conservés entre les mammifères et le poisson zèbre. Différents anticorps utilisés lors de l’établissement des diagnostics cliniques chez l’homme, ont une réactivité croisée avec des antigènes présents chez le poisson zèbre et peuvent donc être utilisés [16].
Les embryons de un jour exercent un mouvement d’enroulement qui permet d’étudier de façon aisée la fonction musculaire dépendante des fibres lentes [15]. Le comportement locomoteur, qui commence à un jour de vie, est quantifiable et facilement mesurable de manière non invasive et reproductible [17] à deux et trois jours de vie, l’embryon ayant dès lors développé la capacité de réponse d’échappement à un stimulus mécanique2. À deux jours de développement, les muscles des embryons de poisson zèbre sont complètement différenciés. Par la suite, la vitesse et la fréquence du patron de nage peuvent être quantifiées. L’organisation des fibres musculaires est, quant à elle, facilement observable et peut être quantifiée en utilisant leur propriété de biréfringence (voir Glossaire). En effet, lorsqu’elles sont examinées sous une lumière polarisée, les fibres qui se détachent - témoins de la dégénérescence - apparaissent plus sombres [18].
L’embryon étant transparent, il est possible d’étudier les muscles squelettiques par immunohistochimie. On peut également utiliser des poissons génétiquement modifiés, exprimant des protéines fluorescentes dont les séquences codantes sont placées sous le contrôle d’un promoteur spécifique des muscles, permettant une étude optique aisée de la structure, mais aussi de la fonction des muscles [19]
Les modèles d’étude générés chez le poisson zèbre
Différentes méthodes permettent de manipuler, chez les poissons zèbres, des gènes d’intérêt afin de reproduire des défauts génétiques observés chez les patients. Les morpholinos représentent une stratégie rapide et transitoire. Ils ont cependant montré leurs limites récemment [20]. Certains projets ambitieux qui reposent sur une technique de criblage génétique direct (voir Glossaire) ont montré l’intérêt de cette méthode pour la création de mutations germinales héritables et récessives, et pour l’identification des phénotypes qui y sont associés. Le criblage génétique inverse ou indirect (voir Glossaire) peut également être utilisé.
Dans les années 1990, un effort conséquent a été réalisé par C. Nüsslein-Volhard et ses collègues afin de caractériser, par criblage génétique, une collection de poissons mutants obtenus par mutagenèse chimique grâce à l’administration d’ENU (N-Nitroso-N-éthylurée) [21]. Parmi les milliers de lignées ainsi produites, certaines présentaient les caractéristiques de maladies neuromusculaires : réduction de la mobilité voire immobilité, peu ou pas de striation des fibres musculaires, dégénérescence musculaire, mais également défauts cardiaques [21]. Dans les protocoles de criblage génétique direct, deux types d’observations visuelles ont été utilisés pour sélectionner rapidement un grand nombre de poissons : le phénomène optique de la biréfringence et les défauts de mobilité [22]. Ces criblages visuels simples peuvent être associés à d’autres mesures de l’intégrité musculaire comme la coloration par le bleu Evans. Injecté dans la larve de poisson, le colorant pénètre uniquement dans les fibres musculaires endommagées, permettant ainsi d’évaluer l’intégrité du sarcolemme (voir Glossaire). Il est également possible d’enregistrer le détachement des myofibres en temps réel [23], permettant l’identification dans les lignées d’un grand nombre de mutations affectant des gènes impliqués dans les myopathies congénitales, mais aussi les dystrophies musculaires et les DMC.
Les techniques de modifications du génome (genome editing), permettant de modifier un gène d’intérêt, ont été plus récemment décrites et sont dénommées criblage génétique indirect [24]. Ce sont des techniques utilisant le système des TALE nucléases [52] (→) et Crispr/Cas9 [24, 53] (→) dont l’efficacité pour générer des mutations est maintenant démontrée. Certaines limitations de ces techniques existent cependant, comme la difficulté de créer des mutations ponctuelles spécifiques ou la génération d’effets hors cibles.
(→) Voir la Synthèse de B. Dupret et P.O. Angrand, m/s n° 2, février 2014, page 186
(→) Voir la Nouvelle de H. Gilgenkrantz, m/s n° 12, décembre 2014, page 1066
Les différentes techniques de criblage ont montré leur intérêt pour l’identification et la création, de manière aisée, de poissons zèbres présentant des mutations ciblées sur des gènes d’intérêt, générant ainsi de nouveaux modèles de DMC.
Les principaux modèles de dystrophies musculaires congénitales chez le poisson
Atteinte des isoformes de laminine : les poissons candyfloss (barbe à papa) et softy (douillet)
On trouve différentes isoformes de la laminine (lam) dans le myotome du poisson zèbre. Ces isoformes sont composées pour la majeure partie des chaînes α2 (lama2) ou α4 (lama4), β2 (lamb2) et γ1 (lamc1) [25]. Le rôle respectif de ces formes de laminine a été établi récemment par l’équipe de P.D. Currie, en utilisant des tests d’épistasie (voir Glossaire) génétiques [26]. L’étude du mutant lama2 du poisson zèbre, appelé candyfloss (caf), révèle un phénotype dystrophique sévère. Chez ce mutant, les fibres musculaires se détachent progressivement de la lame basale sans rupture du sarcolemme. Ce mutant candyfloss présente les principales caractéristiques de la DMC, notamment avec un déficit primaire en mérosine (retrouvé dans la DMC de type 1A, DMC1A) [23] (Tableau I). L’étude réalisée par Hall et ses collègues consistait en l’observation en temps réel du détachement des fibres musculaires de la larve de poisson, élevée dans des conditions anesthésiantes. Après élimination de l’anesthésiant, l’activité motrice des larves mutées traitées, évaluée dans un milieu visqueux (constitué de méthylcellulose) utilisé afin d’augmenter la charge mécanique, reprend et entraîne un processus dystrophique rapide. Cette élégante étude démontre, chez le mutant caf, un détachement de l’extrémité des fibres musculaires au niveau de la jonction myotendineuse (voir Glossaire). La sévérité du détachement est proportionnelle à la charge mécanique appliquée à la larve. Cependant, chez ce mutant, la myogenèse et la fusion des myoblastes sont normales et les motoneurones ne semblent pas affectés. Une autre mutation du gène codant lama2, au niveau d’un site d’épissage, a récemment été mise en évidence par criblage des mutations induites par l’exposition à l’ENU [27] (Tableau I).
Modèles poissons zèbres de dystrophies musculaires congénitales. DG : dystroglycane.
Ce mutant lama2cl501/cl501 présente une altération de la matrice extracellulaire et un détachement des myofibres de la membrane basale.
Chez l’homme, le déficit en lama2 s’accompagne d’anomalies cérébrales comme une hypodensité et une hypomyélinisation de la substance blanche, ainsi que, parfois, des changements structuraux. Chez le poisson mutant lama2cl501/cl501 , la taille du cerveau est anormale, suggérant l’existence d’altérations de processus développementaux semblables chez le poisson zèbre et chez les patients présentant une DMC1A [27, 28].
Un mutant comportant une altération faux sens du gène lamb2 a également été mis en évidence [27] (Tableau I), le mutant softy. Chez ce mutant, la myogenèse est normale, mais un détachement des fibres musculaires se produit chez la larve au niveau du myosepte vertical. Contrairement à ce qui est observé chez le mutant caf, les myofibres détachées ne deviennent pas nécrotiques et forment des myoseptes ectopiques au sein du myotome.
Ces modèles reproduisent les signes pathologiques et les défauts musculaires observés chez les patients. Ce sont donc potentiellement des modèles de choix pour développer des thérapies visant à améliorer la fonction musculaire des patients atteints de DMC [27].
Atteinte des collagènes VI et XXII
Deux DMC se caractérisent par un déficit en collagène VI : la DMC d’Ullrich, qui représente 20 à 30 % des DMC, et la myopathie de Bethlem. Le modèle murin mutant pour le gène col6a1 (collagen, type VI, alpha 1) présente un phénotype de dystrophie de type myopathie de Bethlem de forme légère, mais aucun modèle modèle murin ne permet d’étudier le type Ullrich, une forme plus sévère de la maladie, induite par la délétion du collagène VI [29]. Dans l’étude de Telfer et ses collègues, l’invalidation transitoire de col6a1 chez le poisson zèbre, via des morpholinos antisens, permet de simuler les deux phénotypes de myopathies [21] (Tableau I). Le premier modèle reproduit la forme humaine la plus sévère, la dystrophie musculaire de type Ullrich. Elle s’accompagne d’apoptose et d’une altération de la membrane du sarcolemme avec une anomalie structurale des mitochondries. Le deuxième mime une forme moins sévère de myopathie. Les auteurs ont examiné l’effet d’un inhibiteur de la pompe à protons, la cyclosporine A, sur les conséquences phénotypiques de la mutation. La cyclosporine améliore le comportement locomoteur et l’ultrastructure des organites chez la larve de poisson zèbre mutée. Ceci confirme l’intérêt du modèle dans une perspective thérapeutique de la myopathie d’Ullrich [21]. Des travaux semblables ont été publiés chez la souris et l’homme [30, 31].
D’autres formes de collagène sont importantes pour le maintien de l’attachement des myofibres à la jonction myotendineuse. Un défaut de collagène XXII - créé grâce à un morpholino antisens spécifique du gène col22a1 (collagen, type XXII, alpha 1) - a été récemment étudié chez le poisson. Dans ce modèle, une altération du comportement locomoteur et un phénotype dystrophique ont été observés [32] (Tableau I), faisant de ce gène un nouveau candidat atteint dans certaines DMC chez l’homme. Au cours de cette étude, les auteurs ont montré l’implication de l’intégrine a7/b1, et un rôle pour le collagène XXII dans le renforcement de l’attachement des fibres musculaires à la jonction myotendineuse [32].
Loss contact (perte de contact) et patchytail (queue fragmentée) : atteinte des complexes d’adhésion de la jonction myotendineuse
Les principaux récepteurs de la laminine des muscles squelettiques se situent au niveau de la membrane plasmique [26]. Il s’agit de protéines transmembranaires, le dystroglycane et l’intégrine a7/β1 (Figure 3). Pour maintenir l’intégrité musculaire, l’intégrine α7/β1 et le dystroglycane ont des rôles redondants et complémentaires [33, 34]. L’intégrine α7/β1 est présente notamment au niveau de la jonction myotendineuse. Elle représente un support structural important pour la stabilité du muscle squelettique [35]. Le phénotype dystrophique du mutant loss contact du poisson zèbre résulte d’une mutation du gène codant la kinase associée à l’intégrine, l’ILK (integrin/integrin-linked kinase), qui se lie au domaine intracellulaire de l’intégrine β1 [36] (Tableau I). Le recrutement de cette protéine d’adhésion focale au niveau de la jonction myotendineuse nécessite lama4 (laminin, alpha 4) et l’intégrine α7. Elle est de plus nécessaire au maintien de l’intégrité de l’adhésion du muscle à la jonction myotendineuse [34]. L’action d’ILK et celle du complexe de glycoprotéines associées à la dystrophine sont redondantes. Postel et ses collègues ont montré, par la méthode du double hybride réalisée chez la levure, que l’ILK interagit avec la β-parvine (affixine), et que cette interaction est indispensable à l’adhésion des fibres musculaires [34].
Chez le morphant intégrine a7, Goody et ses collègues ont montré que la nicotinamide riboside kinase 2b, qui interagit avec la partie cytoplasmique de l’intégrine β1, peut réduire le phénotype de dégénérescence musculaire [37]. Ils montrent également que l’intégrine a6 est présente au niveau du myosepte vertical, démontrant que les composants intracellulaires qui interagissent avec les intégrines sont nécessaires à la stabilité mécanique des fibres musculaires au niveau de la jonction myotendineuse, même s’ils ne sont pas indispensables à son assemblage.
La protéine dystroglycane interagit avec lama2 de la lame basale par l’intermédiaire de sa sous-unité α, et avec le cytoplasme via sa sous-unité β. Une des protéines qui lui sont associées, la dystrophine, permet notamment de transférer la force de contraction musculaire à la membrane basale. Le modèle murin développé ne permet pas de mimer la sévérité des symptômes cliniques observés chez l’homme au cours de la myopathie de Duchenne (DMD). En revanche, chez le poisson zèbre, différents allèles de mutations nulles (voir Glossaire) de dystrophine, ou des morpholinos spécifiques, ont permis de montrer que la progression et la sévérité du phénotype chez le poisson étaient proches de celles observées chez les patients atteints de dystrophinopathies [27] (Tableau I). L’absence de dystrophine entraîne l’accumulation progressive de fibres nécrotiques. Elle provoque également une importante fibrose accompagnée d’une inflammation. Chez le poisson zèbre, en conditions anesthésiantes, les myofibres restent attachées. Les fibres ne se détachent que lorsque la larve est replacée dans des conditions normales et que ses premiers mouvements se mettent en place [20]. Le mutant sapje (sap)3 a permis de montrer que la dystrophine est essentielle pour maintenir l’intégrité du sarcolemme au niveau de la jonction myotendineuse [38]. Récemment, il a également été montré que le traitement par la trichostatine A (TSA), un inhibiteur des histone déacétylases (HDAC), pouvait améliorer le processus évolutif de la maladie chez le poisson, ouvrant ainsi des perspectives pour une thérapie fondée sur l’utilisation de cet inhibiteur [39].
La protéine partenaire de la dystrophine, le dystroglycane, a également été largement étudiée chez le poisson zèbre. L’utilisation de morpholinos antisens pour invalider transitoirement le gène codant le dystroglycane engendre des mutants présentant une altération des fibres musculaires avec des signes d’apoptose et de nécrose [8]. dag1hu3072 et patchytail sont deux mutants de poisson zèbre caractérisés par une absence de sous-unités α et β du dystroglycane [32]. Ces mutants ont des phénotypes différents, illustrant la diversité des signes cliniques que l’on peut observer chez des patients atteints de dystrophies musculaires. Chez le mutant dag1hu3072 , les myofibres se détachent du myosepte et le sarcolemme se rompt [27]. Chez le mutant patchytail, le phénotype est plus léger et on n’observe qu’un myosepte plus large [32].
Le complexe d’adhésion à la jonction myotendineuse joue donc un rôle essentiel dans la stabilisation structurale entre le cytosquelette des myofibres et la lame basale. Les mutants obtenus chez le poisson zèbre permettent l’étude de la jonction myotendineuse et des dystrophies qui résultent de son altération.
La glycosylation de l’α-dystroglycane
L’avantage du poisson zèbre dans l’étude des dystroglycanopathies repose sur la conservation, chez ce poisson, des protéines de glycosylation de l’α-dystroglycane [40]. Beaucoup de gènes impliqués dans les dystroglycanopathies sont présents chez le poisson et permettent ainsi l’étude de la pathogenèse de la maladie observée chez l’homme (Tableau I). Contrairement aux modèles murins où on observe une létalité précoce des embryons affectés par ces mutations de l’a-dystroglycane ou de la voie de glycosylation de ce dernier [22], plusieurs signes cliniques sont exprimés chez les poissons qui présentent une mutation de ces mêmes gènes. Des anticorps spécifiques, reconnaissant en western blot ou en immunofluorescence le motif de glycosylation de l’α-dystroglycane, révèlent une hypoglycosylation. Une étude histologique réalisée chez ces poissons a également permis de révéler une désorganisation de la lame basale et un détachement des fibres musculaires au niveau du myosepte vertical. Dans certains modèles, des défauts structuraux de l’œil ou du cerveau ont également été associés au phénotype, reproduisant ainsi la pathogenèse des dystroglycanopathies chez l’homme.
En effet, chez les patients présentant une dystroglycanopathie, la dystrophie musculaire peut être associée à des anomalies cérébro-oculaires. C’est le cas des patients atteints du syndrome de Walker-Warburg, du syndrome muscle-œil-cerveau, de la DMC de Fukuyama ou encore de la dystrophie musculaire précoce avec microcéphalie et retard mental. À ce jour, un défaut génétique n’a pu être mis en évidence que chez seulement la moitié des patients présentant les caractéristiques cliniques des dystroglycanopathies. Le poisson zèbre a permis de caractériser certains des gènes impliqués dans ces atteintes et, à ce jour, quinze mutations de gènes ont été mises en évidence [41]. L’étude de ces maladies repose sur l’analyse histologique des muscles. Elle permet de révèler un défaut de la glycosylation de l’α-dystroglycane, responsable d’une réduction de son affinité pour lama2 au niveau de la membrane basale, ce qui provoque le détachement des fibres musculaires [42].
La contribution du poisson zèbre à l’étude des dystroglycanopathies est donc cruciale. La pléiotropie des gènes identifiés dans ce modèle laisse présager une extension des recherches utilisant ce modèle pour cette classe de DMC.
Conclusion
Des mutations situées au niveau de plus de vingt-cinq gènes ont été identifiées comme responsables de différentes DMC [4]. D’autres restent encore à identifier. Grâce à ses caractéristiques, le poisson zèbre s’est avéré un outil précieux pour appréhender la pathogenèse de ces maladies. Il peut, en outre, contribuer au développement de stratégies thérapeutiques [27]. La modélisation future de nouvelles pathologies musculaires, conduisant à l’identification de nouveaux gènes potentiellement impliqués dans ces maladies [32], ou confirmant l’implication de gènes dont l’altération a été caractérisée chez l’homme [43, 44], sera certainement source de progrès pour l’étude de ces pathologies graves.
Liens d’intérêt
L’auteur déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Glossaire
Biréfringence : propriété d’un matériau dans lequel la lumière se propage de manière anisotrope ; cette propriété s’illustre au sein de l’échantillon observé par une coloration différentielle des tissus et permet de mesurer le niveau de l’atteinte musculaire chez le poisson en microscopie à lumière polarisée.
Criblage génétique direct : approche visant à identifier les gènes responsables d’un phénotype particulier.
Criblage génétique indirect ou inverse : approche permettant de créer des mutations dans un gène donné, dont les phénotypes sont ensuite analysés.
Épistasie : interaction entre deux ou plusieurs gènes contrôlant un même phénotype particulier.
Jonction myotendineuse : site de transfert de la force générée par la contraction du muscle squelettique vers le tendon (Figure 2).
Morpholino : molécule synthétique antisens d’environ 25 bases qui se fixe aux séquences complémentaires d’ARN par appariement de bases. Il peut être utilisé pour bloquer la traduction ou modifier l’épissage du préARNm et ainsi diminuer ou invalider l’expression d’un gène. Un organisme traité par morpholino est appelé un morphant.
Mutation nulle : une mutation nulle élimine complètement la fonction d’un gène.
Myosepte : lame conjonctive séparant les masses musculaires latérales, les myotomes.
Sarcolemme : membrane plasmique des fibres musculaires striées.
Somites : structures embryonnaires situées de part et d’autre du tube neural et de la chorde et qui sont à l’origine, entre autres, des muscles striés.
Fibres musculaires lentes (fibres rouges, type 1) : ces fibres possèdent une forte densité capillaire, une section de petit diamètre, une faible puissance et une forte endurance. Elles utilisent le métabolisme oxydatif et sont donc adaptées aux efforts aérobie. Elles possèdent une vitesse de conduction lente, un seuil d’activation bas (elles sont donc mobilisées pour de faibles contractions), et sont très résistantes à la fatigue (et donc impliquées dans les exercices prolongés).
Fibres musculaires rapides (fibres blanches, type 2) : ces fibres possèdent une faible densité capillaire, une section de grand diamètre, une forte puissance et une faible endurance. Elles utilisent le métabolisme oxydatif et le métabolisme glycolytique, et sont donc adaptées aux efforts anaérobie. Elles possèdent une faible résistance à la fatigue et une force de contraction élevée.
Voir dans m/s un autre exemple de pathologie pour laquelle le danio a permis des progrès importants [51].
Le comportement locomoteur des poissons zèbres peut être quantifié en utilisant le test de touch escape. Le poisson est « touché » (stimulus mécanique) et sa réponse ou son patron d’échappement de nage est enregistré et quantifié.
Le mutant sapje présente une mutation non-sens qui se traduit par un codon stop au niveau du gène de la dystrophine.
Références
- Sparks S, Quijano-Roy S, Harper A, et al. Pagon RA, Adam MP, Ardinger HH, et al. Congenital muscular dystrophy overview. GeneReviews(R) 1993 ; Seattle (WA): University of Washington. [Google Scholar]
- Emery AE. The muscular dystrophies. Lancet 2002 ; 359 : 687–695. [CrossRef] [PubMed] [Google Scholar]
- Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol 2006 ; 7 : 762–773. [CrossRef] [PubMed] [Google Scholar]
- Kaplan JC, Hamroun D. The 2015 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul Disord 2014 ; 24 : 1123–1153. [CrossRef] [PubMed] [Google Scholar]
- Lisi MT, Cohn RD. Congenital muscular dystrophies: new aspects of an expanding group of disorders. Biochem Biophys Acta 2007 ; 1772 : 159–172. [Google Scholar]
- Santoriello C, Zon LI. Hooked! Modeling human disease in zebrafish. J Clin Invest 2012 ; 122 : 2337–2343. [CrossRef] [PubMed] [Google Scholar]
- Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nat Rev Genet 2007 ; 8 : 353–367. [CrossRef] [PubMed] [Google Scholar]
- Berger J, Berger S, Hall TE, et al. Dystrophin-deficient zebrafish feature aspects of the Duchenne muscular dystrophy pathology. Neuromuscul Disord 2010 ; 20 : 826–832. [CrossRef] [PubMed] [Google Scholar]
- Telfer WR, Busta AS, Bonnemann CG, et al. Zebrafish models of collagen VI-related myopathies. Hum Mol Genet 2010 ; 19 : 2433–2444. [CrossRef] [PubMed] [Google Scholar]
- Sicinski P, Geng Y, Ryder-Cook AS, et al. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science 1989 ; 244 : 1578–1580. [CrossRef] [PubMed] [Google Scholar]
- Bassett DI, Bryson-Richardson RJ, Daggett DF, et al. Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development 2003 ; 130 : 5851–5860. [CrossRef] [PubMed] [Google Scholar]
- Jackson HE, Ingham PW. Control of muscle fibre-type diversity during embryonic development: the zebrafish paradigm. Mech Dev 2013 ; 130 : 447–457. [CrossRef] [PubMed] [Google Scholar]
- Dou Y, Andersson-Lendahl M, Arner A. Structure and function of skeletal muscle in zebrafish early larvae. J Gen Physiol 2008 ; 131 : 445–453. [CrossRef] [PubMed] [Google Scholar]
- Guyon JR, Mosley AN, Zhou Y, et al. The dystrophin associated protein complex in zebrafish. Hum Mol Genet 2003 ; 12 : 601–615. [CrossRef] [PubMed] [Google Scholar]
- Parsons MJ, Campos I, Hirst EM, Stemple DL. Removal of dystroglycan causes severe muscular dystrophy in zebrafish embryos. Development 2002 ; 129 : 3505–3512. [PubMed] [Google Scholar]
- Berger J, Currie PD. Zebrafish models flex their muscles to shed light on muscular dystrophies. Dis Mod Mech 2012 ; 5 : 726–732. [CrossRef] [Google Scholar]
- Saint-Amant L, Drapeau P. Time course of the development of motor behaviors in the zebrafish embryo. J Neurobiol 1998 ; 37 : 622–632. [CrossRef] [PubMed] [Google Scholar]
- Granato M, van Eeden FJ, Schach U, et al. Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development 1996 ; 123 : 399–413. [PubMed] [Google Scholar]
- Roostalu U, Strahle U. In vivo imaging of molecular interactions at damaged sarcolemma. Dev Cell 2012 ; 22 : 515–529. [CrossRef] [PubMed] [Google Scholar]
- Schulte-Merker S, Stainier DY. Out with the old, in with the new: reassessing morpholino knockdowns in light of genome editing technology. Development 2014 ; 141 : 3103–3104. [CrossRef] [PubMed] [Google Scholar]
- Haffter P, Granato M, Brand M, et al. The identification of genes with unique and essential functions in the development of the zebrafish. Danio rerio. Development 1996 ; 123 : 1–36. [Google Scholar]
- Gibbs EM, Horstick EJ, Dowling JJ. Swimming into prominence: the zebrafish as a valuable tool for studying human myopathies and muscular dystrophies. FEBS J 2013 ; 280 : 4187–4197. [CrossRef] [PubMed] [Google Scholar]
- Hall TE, Bryson-Richardson RJ, Berger S, et al. The zebrafish candyfloss mutant implicates extracellular matrix adhesion failure in laminin alpha2-deficient congenital muscular dystrophy. Proc Natl Acad Sci USA 2007 ; 104 : 7092–7097. [CrossRef] [Google Scholar]
- Auer TO, Del Bene F. CRISPR/Cas9 and TALEN-mediated knock-in approaches in zebrafish. Methods 2014 ; 69 : 142–150. [CrossRef] [PubMed] [Google Scholar]
- Sztal T, Berger S, Currie PD, Hall TE. Characterization of the laminin gene family and evolution in zebrafish. Dev Dyn 2011 ; 240 : 422–431. [CrossRef] [PubMed] [Google Scholar]
- Sztal TE, Sonntag C, Hall TE, Currie PD. Epistatic dissection of laminin-receptor interactions in dystrophic zebrafish muscle. Hum Mol Genet 2012 ; 21 : 4718–4731. [CrossRef] [PubMed] [Google Scholar]
- Wood AJ, Currie PD. Analysing regenerative potential in zebrafish models of congenital muscular dystrophy. Int J Biochem Cell Biol 2014; 56C : 30–37. [CrossRef] [Google Scholar]
- Gupta VA, Kawahara G, Myers JA, et al. A splice site mutation in laminin-alpha2 results in a severe muscular dystrophy and growth abnormalities in zebrafish. PloS One 2012 ; 7 : e43794. [CrossRef] [PubMed] [Google Scholar]
- Bonaldo P, Braghetta P, Zanetti M, et al. Collagen VI deficiency induces early onset myopathy in the mouse: an animal model for Bethlem myopathy. Hum Mol Genet 1998 ; 7 : 2135–2140. [CrossRef] [PubMed] [Google Scholar]
- Millay DP, Sargent MA, Osinska H, et al. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med 2008 ; 14 : 442–447. [CrossRef] [PubMed] [Google Scholar]
- Merlini L, Angelin A, Tiepolo T, et al. Cyclosporin A corrects mitochondrial dysfunction and muscle apoptosis in patients with collagen VI myopathies. Proc Natl Acad Sci USA 2008 ; 105 : 5225–5229. [CrossRef] [Google Scholar]
- Charvet B, Guiraud A, Malbouyres M, et al. Knockdown of col22a1 gene in zebrafish induces a muscular dystrophy by disruption of the myotendinous junction. Development 2013 ; 140 : 4602–4613. [CrossRef] [PubMed] [Google Scholar]
- Rooney JE, Welser JV, Dechert MA, et al. Severe muscular dystrophy in mice that lack dystrophin and alpha7 integrin. J Cell Sci 2006 ; 119 : 2185–2195. [CrossRef] [PubMed] [Google Scholar]
- Jimenez-Mallebrera C, Brown SC, Sewry CA, Muntoni F. Congenital muscular dystrophy: molecular and cellular aspects. Cell Mol Life Sci 2005 ; 62 : 809–823. [CrossRef] [PubMed] [Google Scholar]
- Danen EH, Sonnenberg A. Integrins in regulation of tissue development and function. J Pathol 2003 ; 200 : 471–480. [CrossRef] [PubMed] [Google Scholar]
- Lin YY. Muscle diseases in the zebrafish. Neuromusc Dis 2012 ; 22 : 673–684. [CrossRef] [Google Scholar]
- Goody MF, Kelly MW, Reynolds CJ, et al. NAD+ biosynthesis ameliorates a zebrafish model of muscular dystrophy. PLoS Biol 2012 ; 10 : e1001409. [CrossRef] [PubMed] [Google Scholar]
- Gupta V, Kawahara G, Gundry SR, et al. The zebrafish dag1 mutant: a novel genetic model for dystroglycanopathies. Hum Mol Genet 2011 ; 20 : 1712–1725. [CrossRef] [PubMed] [Google Scholar]
- Johnson NM, Farr GH 3rd, Maves L. The HDAC inhibitor TSA ameliorates a zebrafish model of Duchenne muscular dystrophy. PLoS Curr 2013; 5. [Google Scholar]
- Moore CJ, Goh HT, Hewitt JE. Genes required for functional glycosylation of dystroglycan are conserved in zebrafish. Genomics 2008 ; 92 : 159–167. [CrossRef] [PubMed] [Google Scholar]
- Muntoni F, Torelli S, Wells DJ, Brown SC. Muscular dystrophies due to glycosylation defects: diagnosis and therapeutic strategies. Curr Opin Neurol 2011 ; 24 : 437–442. [CrossRef] [PubMed] [Google Scholar]
- Godfrey C, Foley AR, Clement E, Muntoni F. Dystroglycanopathies: coming into focus. Curr Opin Genet Dev 2011 ; 21 : 278–285. [CrossRef] [PubMed] [Google Scholar]
- Stevens E, Carss KJ, Cirak S, et al. Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of alpha-dystroglycan. Am J Hum Genet 2013 ; 92 : 354–365. [CrossRef] [PubMed] [Google Scholar]
- Buysse K, Riemersma M, Powell G, et al. Missense mutations in beta-1,3-N-acetylglucosaminyltransferase 1 (B3GNT1) cause Walker-Warburg syndrome. Hum Mol Genet 2013 ; 22 : 1746–1754. [CrossRef] [PubMed] [Google Scholar]
- Carss KJ, Stevens E, Foley AR, et al. Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of alpha-dystroglycan. Am J Hum Genet 2013 ; 93 : 29–41. [CrossRef] [PubMed] [Google Scholar]
- Manzini MC, Tambunan DE, Hill RS, et al. Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome. Am J Hum Genet 2012 ; 91 : 541–547. [CrossRef] [PubMed] [Google Scholar]
- Roscioli T, Kamsteeg EJ, Buysse K, et al. Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of alpha-dystroglycan. Nat Genet 2012 ; 44 : 581–585. [CrossRef] [PubMed] [Google Scholar]
- Di Costanzo S, Balasubramanian A, Pond HL, et al. POMK mutations disrupt muscle development leading to a spectrum of neuromuscular presentations. Hum Mol Genet 2014 ; 23 : 5781–5792. [CrossRef] [PubMed] [Google Scholar]
- Willer T, Prados B, Falcon-Perez JM, et al. Targeted disruption of the Walker-Warburg syndrome gene Pomt1 in mouse results in embryonic lethality. Proc Natl Acad Sci USA 2004 ; 101 : 14126–14131. [CrossRef] [Google Scholar]
- Steffen LS, Guyon JR, Vogel ED, et al. The zebrafish runzel muscular dystrophy is linked to the titin gene. Dev Biol 2007 ; 309 : 180–192. [CrossRef] [PubMed] [Google Scholar]
- Bernut A, Lutfalla G, Kremer L. Regard à travers le danio pour mieux comprendre les interactions hôte/pathogène. Med Sci (Paris) 2015 ; 31 : 638–646. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Dupret B, Angrand PO. L’ingénierie des génomes par les TALEN. Med Sci (Paris) 2014 ; 30 : 186–193. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- La Gilgenkrantz H.. révolution des CRISPR est en marche. Med Sci (Paris) 2014 ; 30 : 1066–1069. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des tableaux
Modèles poissons zèbres de dystrophies musculaires congénitales. DG : dystroglycane.
Liste des figures
|
Figure 1. Schéma d’un embryon de poisson zèbre deux jours après fécondation. . Lorsqu’un processus dystrophique apparaît, la stabilité structurale de l’ancrage des myofibres est affectée et les fibres se détachent du myosepte vertical. |
| Dans le texte | |
|
Figure 2. Schéma simplifié des complexes d’adhésion de la jonction myotendineuse . Les complexes d’adh販on permettent de relier le cytosquelette d’actine des myofibres à la lame basale. Ces différents complexes se compensent l’un l’autre. CaV3 : caveolin-3 ; ILK : integrin-linked kinase. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.