")
")
| Issue |
Med Sci (Paris)
Volume 28, Number 10, Octobre 2012
|
|
|---|---|---|
| Page(s) | 876 - 882 | |
| Section | Récepteurs couplés aux protéines G | |
| DOI | https://doi.org/10.1051/medsci/20122810017 | |
| Published online | 12 octobre 2012 | |
Les récepteurs couplés aux protéines G dans la lumière
G protein-coupled receptors in the spotlight
1
Institut de génomique fonctionnelle, département de pharmacologie moléculaire, UMR 5203 CNRS, Inserm U661, universités de Montpellier I et II, 141 rue de La Cardonille, 34094 Montpellier, France
2
Medical research council (MRC), laboratory of molecular biology, Hills Road, Cambridge CB2 0QH, Royaume-Uni
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
**
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les récepteurs couplés aux protéines G (RCPG) représentent la plus grande famille de protéines membranaires. Localisés à la surface des cellules dans la membrane plasmique, les RCPG se lient à une grande diversité de ligands tels que les hormones, les nucléotides, les lipides, les ions et les neurotransmetteurs, et sont des intermédiaires clés dans la transmission de signaux de l’extérieur vers l’intérieur de la cellule. Durant les cinq dernières années, d’énormes progrès ont été réalisés dans le domaine de la cristallographie des RCPG. Les structures tridimensionnelles des récepteurs β1- et β2-adrénergique (β1-AR et β2-AR) et du récepteur de l’adénosine (A2AR) complexés à des agonistes en sont une illustration, avec notamment la publication de la structure du récepteur β2-AR complexé à la protéine G stimulatrice (Gs). Cette synthèse présente une vue d’ensemble des structures tridimensionnelles des RCPG et de leur mécanisme d’activation.
Abstract
G protein-coupled receptors (GPCR) are the largest family of integral membrane proteins found in the plasma membrane of mammalian cells. GPCR respond to a large variety of ligands such as amines, lipids, hormones and amino-acids, which are involved in inter-cellular signalling events in a multitude of physiological and pathological processes. GPCRs are therefore key regulators of signal transduction by which cells respond to variations in their environment. During the last five years, striking progress has been made to solve high-resolution structure of GPCR. The most recent successes are the structures of the β1 and β2 adrenoreceptors and the adenosine A2A receptor bound to a variety of agonists. Most importantly, the structure of the β2 adrenoreceptor in complex with a trimeric G protein, Gs, was recently reported. This review will present an overview of the X-ray structure determination of the GPCR and of their activation mechanism.
© 2012 médecine/sciences – Inserm / SRMS
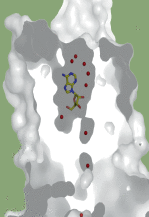
Photo : adénosine (© Guillaume Lebon).
Les récepteurs couplés aux protéines G (RCPG) constituent la plus grande famille de protéines membranaires. Chez l’homme, environ 800 gènes codent pour des RCPG [1]. Ces récepteurs, exprimés dans tous les organes du corps humain, sont impliqués dans les processus physiologiques importants et, par conséquent, dans les dysfonctionnements pathologiques de l’organisme. Ils représentent donc des cibles thérapeutiques très intéressantes pour le traitement des maladies cardiaques, des désordres inflammatoires ou du système nerveux central. Les RCPG ont de très nombreux ligands : des hormones, nucléotides, lipides, ions et neurotransmetteurs qui, en se fixant sur ces récepteurs, induisent un changement de leur conformation, permettant ainsi la transmission d’un signal extracellulaire vers l’intérieur de la cellule. L’activation des RCPG est alors convertie en signal cellulaire via l’activation des protéines G et d’autres effecteurs cellulaires comme les protéines arrestines [2, 3].
L’activité des RCPG est modulée par diverses catégories de ligands qui induisent et stabilisent différentes conformations du récepteur. Les agonistes activent le récepteur et le stabilisent préférentiellement dans une conformation active (R*) ; c’est le cas de l’adénosine pour le récepteur de l’adénosine. Les antagonistes limitent l’activité du récepteur à son activité basale, qui correspond à l’activité du récepteur en absence de stimulation par un agoniste. Un agoniste inverse, quant à lui, inactive totalement le récepteur et, ainsi, inhibe même son activité basale. Antagonistes et agonistes inverses stabilisent donc préférentiellement une conformation similaire, inactive, des récepteurs (R) [4].
Les RCPG interagissent avec différents partenaires intracellulaires tels que les protéines G ou les protéines arrestines. Chaque partenaire protéique va ainsi potentiellement stabiliser des conformations spécifiques du récepteur en interagissant avec le versant intracellulaire de ce dernier. Si l’on considère la grande diversité de ligands et de voies de signalisation qui peuvent être activées par un même récepteur, il apparaît évident que les RCPG sont des protéines très modulables pouvant adopter de multiples conformations [3]. Cette flexibilité peut être représentée par un large éventail d’états conformationnels définis par la fixation d’un ligand sur la surface extracellulaire du récepteur, mais aussi par l’interaction du récepteur avec des effecteurs cellulaires. Ces différents états conformationnels ont été étudiés par différentes approches telles que la mutagenèse dirigée, la modélisation moléculaire, ou encore les approches biophysiques. Cependant, la flexibilité conformationnelle des RCPG a longtemps limité leur analyse par des approches structurales telles que la cristallographie aux rayons X. De récents succès dans le domaine de la cristallisation des RCPG ont permis toutefois d’étudier la diversité structurale des RCPG de classe A et d’approfondir les connaissances sur leur mécanisme d’activation et de couplage aux protéines G [5–7]. Ces nouvelles avancées ont aussi permis de caractériser au niveau moléculaire les conformations inactive R et active R*. Cette revue présente l’état des connaissances sur les structures cristallographiques des RCPG de classe A, avec un intérêt particulier pour les récepteurs β1-adrénergique (β1-AR) de la dinde, β2-adrénergique (β2-AR) humain et le récepteur de l’adénosine (A2AR). En effet, l’analyse des structures des β1-AR, β2-AR et A2AR, suggère la conservation d’un mécanisme d’activation commun pour ces trois récepteurs de classe A.
La cristallogenèse des RCPG
La première structure cristallographique d’un RCPG de classe A publiée est celle de la rhodopsine, une protéine activable par la lumière. La protéine rhodopsine est un modèle de référence pour l’étude structurale des RCPG. Les structures cristallographiques de haute résolution de la rhodopsine ont fait l’objet d’excellentes revues [6, 8] et ne seront que très brièvement mentionnées ici.
Depuis 2007, l’étude structurale par cristallographie aux rayons X des RCPG connaît un essor formidable. En effet, il a fallu attendre sept ans après la publication de la structure de la protéine rhodopsine, pour découvrir la première structure tridimensionnelle d’un RCPG liant une hormone, le β2-AR [9]. Cette structure a été obtenue en utilisant un anticorps (Fab5) dirigé contre la 3e boucle intracellulaire. La cristallogenèse des RCPG a longtemps été limitée par la difficulté à produire des cristaux de qualité diffractant à haute résolution [10]. En effet, l’extraction des récepteurs de la membrane cellulaire à l’aide de détergents est une étape délicate qui requiert l’utilisation de composés additifs (comme par exemple le cholestérol) pour améliorer la stabilité du récepteur et ainsi préserver sa structure tridimensionnelle [11, 43] (→). La flexibilité de la conformation des RCPG représente un obstacle supplémentaire pour l’obtention de cristaux de qualité.
(→) Voir la synthèse de J.L. Banères et B. Mouillac, page 837 de ce numéro
Différentes approches ont ainsi été développées afin de résoudre ces difficultés. Une première stratégie consiste à fusionner la protéine du lysozyme T4 (T4L) à l’extrémité intracellulaire des hélices 5 and 6, en remplacement de la boucle ICL3 (intracellular loop 3) (Figure 1) [12]. La protéine de fusion T4L permet d’augmenter la surface hydrophile du récepteur solubilisé à l’aide de détergents, tout en réduisant sa flexibilité et en favorisant ainsi la formation du cristal. Cette technique a permis de résoudre la structure haute résolution du récepteur β2-AR humain lié à l’antagoniste carazolol [13] ou à divers antagonistes et agonistes inverses [11, 14]. Cette approche a ensuite été utilisée pour cristalliser et résoudre la structure tridimensionnelle du récepteur de l’adénosine (A2A) [15], de la dopamine (D3) [16], de l’histamine (H1) [17], le récepteur des chémokines CXCR4 (chemokine [C-X-C motif]receptor 4) [18], les récepteurs muscariniques M2 et M3 [19, 20], le récepteur des sphingosine-1-phosphate (S1P1) [21] (Tableau I) et les récepteurs mu et delta des opiacés [42] (→), tous complexés à des antagonistes ou agonistes inverses, représentant ainsi une conformation inactive R. Une seconde stratégie, basée sur l’introduction de mutations ponctuelles et dirigées, permet d’améliorer dans le temps la stabilité du récepteur solubilisé : la thermostabilisation par mutagenèse dirigée [22–25]. Cette approche a permis de résoudre la structure tridimensionnelle du β1-AR de la dinde lié à un antagoniste (cyanopindolol), et la structure du A2AR lié à différents antagonistes (dont son antagoniste naturel, la caféine) (Figure 1) [26–28].
(→) Voir aussi la synthèse de S. Granier, page 870 de ce numéro
 |
Figure 1. Illustration des différentes stratégies utilisées pour l’étude cristallographique des RCPG. A. Structure du β1-AR thermostable de la dinde (2VT4) lié à l’antagoniste cyanopindolol (vert). Les mutations thermostabilisantes sont représentées en violet. B. Structure du β2-AR-T4L complexé à l’antagoniste carazolol (jaune) (2RH1). La protéine T4L (cyan) a été fusionnée aux extrémités intracellulaires des hélices 5 et 6. C. Structure du β2-AR-T4L lié à l’agoniste BI-167107 en complexe avec un anticorps de lama nb80 (bleu marine) (3POG). D. Structure du β2-AR-T4L lié à l’agoniste BI-167107 (jaune) en complexe avec la protéine G trimérique (α en rouge, β en jaune et γ en rose) (3NS6). L’anticorps de lama nb35 (violet) stabilise la sous-unité Gαs. La protéine de fusion T4L (cyan) a été introduite à l’extrémité amino-terminale du récepteur. |
Structures des RCPG liant des ligands diffusibles. PDB : protein data bank.
La cristallisation d’un RCPG dans sa conformation active - lié à un agoniste - est plus difficile à obtenir que celle d’un RCPG dans sa conformation inactive. La conformation active des RCPG est énergétiquement plus instable et donc plus difficile à caractériser. L’année 2011 a cependant été riche en succès. La première conformation active (R*) du β2-AR humain a été obtenue dans un premier temps à l’aide d’un anticorps de lama (nb80) (Figure 1) [29, 30]. Cet anticorps de lama a été sélectionné parce qu’il se lie au récepteur et mime la fixation de la protéine G. Dans le même temps, la structure tridimensionnelle du β2-AR-T4L lié de manière covalente à un agoniste, FAUC50, et celle du β1-AR thermostable de la dinde lié à différents agonistes (tels que l’isoprénaline, un agoniste non sélectif des β-AR) ont été résolues [31, 32]. Il est à noter que la conformation active de la protéine rhodopsine, ainsi que la structure du mutant constitutivement actif E113Q, toutes deux publiées récemment, complètent les connaissances structurales de la conformation active de l’opsine1 [33–35]. C’est en utilisant la stratégie de thermostabilisation que nous avons pu résoudre la structure du A2AR humain lié à son agoniste naturel (adénosine) [36, 37]. Dans le même temps, l’utilisation de la protéine de fusion T4L a permis de cocristalliser le A2AR avec le ligand UK-432097, ligand développé pour le traitement de la bronchopneumopathie pulmonaire obstructive chronique [38]. Plus récemment, la structure du β2-AR lié à la protéine G sous sa forme trimérique a été publiée, dévoilant ainsi la conformation active (R*) du β2-AR (Figure 1) [39]. Cette dernière structure a été obtenue en utilisant à la fois la protéine de fusion T4L fusionnée à l’extrémité amino-terminale du récepteur et un anticorps de lama (nb35) qui réduit la flexibilité de la sous-unité α de la protéine G (Figure 1). Nous disposons maintenant de structures tridimensionnelles représentant différents états conformationnels des RCPG, inactifs R ou actifs R*. La comparaison de ces différentes structures révèle des similitudes et des différences au niveau des récepteurs de classe A.
La structure des RCPG
La résolution des structures tridimensionnelles des RCPG confirme la prédiction d’une architecture commune, composée de sept hélices transmembranaires (TM1 à TM7) et d’une 8e hélice (H8) positionnée parallèlement à la surface interne de la membrane plasmique. Seule la structure du récepteur CXCR4 est dépourvue de cette hélice supplémentaire [18]. Les 7 hélices transmembranaires sont reliées entre elles par trois boucles extracellulaires (ECL1, 2 et 3 reliant respectivement les TM2 et 3, TM4 et 5, TM6 et 7) et trois boucles intracellulaires (ICL1, 2 et 3 reliant respectivement les TM1 et 2, TM3 et 4, TM5 et 6). Les boucles intra- et extracellulaires sont de tailles différentes suivant les récepteurs et sont le reflet d’importantes variabilités structurales des RCPG, en particulier la boucle ECL2. Dans le cas de la rhodopsine, la boucle ECL2 forme un clapet qui se referme sur la poche de fixation du rétinal. Le long domaine amino-terminal complète cette organisation moléculaire et renforce la stabilité du complexe ainsi formé [40]. Le site de fixation est beaucoup plus exposé au solvant dans le cas du récepteur liant des ligands diffusibles. Dans le cas des récepteurs β1-AR, β2-AR et A2AR, ECL2 se structure en une courte hélice a (environ deux tours d’hélice) et peut jouer des rôles différents. Pour les β1-AR et β2-AR, la boucle ECL2 participe à la formation du site de fixation du ligand, alors que pour l’A2AR, le résidu F168 localisé dans cette boucle est indispensable à la fixation de l’adénosine, et donc à la fonctionnalité du récepteur [12, 31, 36].
Il existe une grande diversité dans les acides aminés qui composent la poche de fixation des ligands des RCPG. Cette diversité est le reflet du grand nombre de molécules, neurotransmetteurs ou hormones qui se lient spécifiquement à un seul type de récepteur, que ce soit un récepteur aminergique (β-AR, dopaminergique), nucléotidique (récepteur de l’adénosine) ou tout autre type de récepteur. Cependant, il est classique qu’entre sous-types de récepteurs qui répondent à un même ligand, comme par exemple les récepteurs de l’adénosine A1, A2A, A2B et A3, il existe un haut degré de conservation pour les résidus directement impliqués dans la fixation du ligand. Sur 16 résidus en contact direct avec l’adénosine dans la structure du A2AR humain, 15 sont strictement conservés pour les sous-types A1 et A2B, et 10 pour le sous-type A3 [36]. Il en est de même pour les β1-AR et β2-AR [7, 31]. La spécificité de fixation des ligands pour un sous-type impliquera davantage les résidus localisés dans les boucles extracellulaires, sources de la plus grande diversité entre les RCPG.
Afin d’activer la protéine G, la liaison d’un agoniste dans le site de fixation d’un RCPG induit des changements conformationnels correspondant aux mouvements des différents TM. Ces changements conformationnels sont de petite amplitude au niveau de la poche de fixation de l’agoniste, et c’est au niveau intracellulaire que l’on peut observer des modifications plus spectaculaires.
Mécanisme d’activation des RCPG
Nous disposons de multiples structures de RCPG liées à leur agoniste, comme par exemple le β1-AR, β2-AR, A2AR ou encore la rhodopsine [6]. Les structures actives R* du β2-AR, liées soit à un anticorps de lama (nb80), soit à la protéine trimérique Gs, sont considérées comme représentatives de la conformation active d’un RCPG [29, 39]. La présence de la protéine G ou de l’anticorps de lama (nb80) est indispensable à la stabilisation de la conformation active R*. La liaison de l’agoniste seul ne permet pas de stabiliser la conformation active R*. En effet, les structures du β1-AR lié aux différents agonistes et du β2-AR lié de manière covalente à un agoniste sont très similaires à la conformation inactive R. Les structures du A2AR lié à l’adénosine ou encore au composé UK-432097 représentent des conformations très similaires à la conformation active R* du récepteur β2-AR. Ces conformations ont été proposées comme représentatives d’un état intermédiaire entre les conformations inactive R et active R* [36]. Les différentes structures des β1-AR, β2-AR et A2AR suggèrent des changements de conformation et un mécanisme d’activation conservés entre ces différents récepteurs, ainsi que l’existence d’une conformation active R* similaire. Par ailleurs, les β1-AR, β2-AR et A2AR interagissent tous avec la même protéine Gs. Cependant, il est probable que les conformations actives des RCPG expriment un degré de variabilité afin de rendre compte de la diversité des protéines G pouvant interagir avec ces récepteurs.
Dans le cas des récepteurs de classe A (β1-AR, β2-AR et A2AR), la fixation d’un agoniste induit la contraction de la poche de fixation du récepteur d’environ 1 Å [29, 31, 36]. Cette contraction représente la première étape de l’activation du récepteur. Les structures représentant les conformations R* des β2-AR et A2AR montrent que les extrémités intracellulaires des TM3 et TM7 et l’extrémité extracellulaire de TM5 sont repositionnées vers l’intérieur du récepteur (Figure 2) [29, 36]. Ces différents réarrangements locaux sont accompagnés d’un mouvement des extrémités intracellulaires des TM5 et TM6, notamment via une rotation de TM6, qui, en s’écartant du récepteur, ouvre ainsi une cavité dans laquelle l’extrémité carboxy-terminale de la sous-unité α de la protéine G vient s’arrimer (Figure 2). Le mouvement de l’hélice 6 est variable, allant de 5 Å dans le cas de la structure du A2AR lié à l’adénosine, jusqu’à 14 Å pour la structure du β2-AR lié à la protéine Gs [29, 36, 39]. Le mouvement de 5 Å de l’hélice 6 dans la structure du A2AR lié à l’adénosine n’est cependant pas suffisant pour permettre l’arrimage de la protéine G, qui est nécessaire à la stabilisation de la conformation active R*. La publication de la structure du β2-AR complexé à la protéine trimérique Gs (α, β et γ) nous montre que le domaine Ras de la sous-unité Gα (GαRas) est principalement responsable de l’interaction entre le β2-AR et la protéine G [39]. L’hélice α5 de la sous-unité alpha de la protéine Gs interagit avec le β2-AR au niveau de l’arginine R3.50 (TM3) du motif conservé E/DRY. Cette interaction est stabilisée par la tyrosine Y7.53 (TM7) appartenant au motif NPxxY, confirmant ainsi l’importance de ces deux motifs conservés (E/DRY et NPxxY) pour le couplage des RCPG avec la protéine Gs [39, 41].
 |
Figure 2. Conformation active R* du récepteur β 2-AR humain. A. Superposition de la structure inactive R (β2-AR lié à l’antagoniste carazolol [2HR1]) et de la conformation active R* (β2-AR complexé à l’agoniste BI-167107 et à la protéine trimérique Gs [3NS6]). Le repositionnement de l’hélice 6 ouvre une cavité dans laquelle la sous-unité Gαs s’arrime. B. Vue de la surface intracellulaire du β2-AR. Le TM5, et surtout le TM6, sont repositionnées vers l’extérieur du récepteur, alors que le TM3 et le TM7 se déplacent vers l’intérieur du récepteur. C. Vue de la surface extracellulaire du β2-AR. Le repositionnement du TM5 vers l’intérieur du récepteur s’effectue au niveau du site de fixation de l’agoniste BI-167107. |
Enfin, pour activer le β2-AR, le ligand BI-167107 établit des interactions au niveau des résidus sérines 203 (S5.42) et 207 (S5.46) localisés dans le TM5 du β2-AR [29]. Il en est de même pour l’isoprénaline, agoniste non spécifique du β-AR, qui active le β1-AR de la dinde en établissant des liaisons hydrogène avec les résidus sérine 211 (S5.42) et 215 (S5.46) [31]. L’adénosine va, quant à elle, établir des liaisons spécifiques impliquant la sérine 277 (S7.42) et l’histidine 278 (H7.43) localisées dans le TM7 du A2AR [36]. Il est intéressant de constater que deux agonistes, BI-167107 pour le β2-AR et l’adénosine pour le A2AR, interagissent avec le récepteur via des contacts qui leur sont spécifiques et différents. Ils induiront in fine des changements conformationnels similaires proposés comme caractéristiques de la conformation active R* des β2-AR et A2AR, et surtout considérés comme nécessaires à la stabilisation de la conformation active R*.
Les études structurales des RCPG par cristallographie aux rayons X ont permis d’approfondir les connaissances sur cette catégorie de récepteurs (en élucidant les modes de fixation des ligands sur les différents types de récepteurs), d’identifier des signatures moléculaires des conformations inactives R et actives R*, et de révéler la diversité structurale de cette classe de protéines membranaires importantes. Ces informations structurales devraient permettre de développer des composés sélectifs d’un type de récepteur et de moduler son activité en fonction des besoins et de l’intérêt thérapeutique du récepteur ciblé. Il est cependant évident que d’autres techniques, comme la résonnance magnétique nucléaire ou encore les expériences de simulation de la dynamique moléculaire in silico, permettront d’approfondir les connaissances et la compréhension des complexes de signalisation formés par les RCPG avec les différents effecteurs cellulaires, comme les protéines G et les arrestines. L’ère de la biologie structurale des RCPG vient de débuter.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Les opsines constituent une famille de protéines qui, grâce à leur liaison au rétinal ou à l’un de ses dérivés, forment la protéine mixte appelée rhodopsine.
Références
- Gloriam DE, Fredriksson R, Schioth HB. The G protein-coupled receptor subset of the rat genome. BMC Genomics 2007 ; 8 : 338. [CrossRef] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol 2007 ; 69 : 483–510. [CrossRef] [PubMed] [Google Scholar]
- Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci 2007 ; 28 : 397–406. [CrossRef] [PubMed] [Google Scholar]
- Dror RO, Arlow DH, Borhani DW, et al. Identification of two distinct inactive conformations of the beta2-adrenergic receptor reconciles structural and biochemical observations. Proc Natl Acad Sci USA 2009 ; 106 : 4689–4694. [CrossRef] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol Sci 2012 ; 33 : 17–27. [CrossRef] [PubMed] [Google Scholar]
- Deupi X, Standfuss J. Structural insights into agonist-induced activation of G-protein-coupled receptors. Curr Opin Struct Biol 2011 ; 21 : 541–551. [CrossRef] [PubMed] [Google Scholar]
- Kobilka BK. Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol Sci 2011 ; 32 : 213–218. [CrossRef] [PubMed] [Google Scholar]
- Choe HW, Park JH, Kim YJ, Ernst OP. Transmembrane signaling by GPCRs: insight from rhodopsin and opsin structures. Neuropharmacology 2011 ; 60 : 52–57. [CrossRef] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Rosenbaum DM, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 2007 ; 450 : 383–387. [CrossRef] [PubMed] [Google Scholar]
- Tate CG, Schertler GF. Engineering G protein-coupled receptors to facilitate their structure determination. Curr Opin Struct Biol 2009 ; 19 : 386–395. [CrossRef] [PubMed] [Google Scholar]
- Hanson MA, Cherezov V, Griffith MT, et al. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure 2008 ; 16 : 897–905. [CrossRef] [PubMed] [Google Scholar]
- Rosenbaum DM, Cherezov V, Hanson MA, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 2007 ; 318 : 1266–1273. [CrossRef] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007 ; 318 : 1258–1265. [CrossRef] [PubMed] [Google Scholar]
- Wacker D, Fenalti G, Brown MA, et al. Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J Am Chem Soc 2010 ; 132 : 11443–11445. [CrossRef] [PubMed] [Google Scholar]
- Jaakola VP, Griffith MT, Hanson MA, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008 ; 322 : 1211–1217. [CrossRef] [PubMed] [Google Scholar]
- Chien EY, Liu W, Zhao Q, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010 ; 330 : 1091–1095. [CrossRef] [PubMed] [Google Scholar]
- Shimamura T, Shiroishi M, Weyand S, et al. Structure of the human histamine H1 receptor complex with doxepin. Nature 2011 ; 475 : 65–70. [CrossRef] [PubMed] [Google Scholar]
- Wu B, Chien EY, Mol CD, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010 ; 330 : 1066–1071. [CrossRef] [PubMed] [Google Scholar]
- Kruse AC, Hu J, Pan AC, et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 2012 ; 482 : 552–556. [CrossRef] [PubMed] [Google Scholar]
- Haga K, Kruse AC, Asada H, et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 2012 ; 482 : 547–551. [CrossRef] [PubMed] [Google Scholar]
- Hanson MA, Roth CB, Jo E, et al. Crystal structure of a lipid G protein-coupled receptor. Science 2012 ; 335 : 851–855. [CrossRef] [PubMed] [Google Scholar]
- Serrano-Vega MJ, Magnani F, Shibata Y, Tate CG. Conformational thermostabilization of the beta1-adrenergic receptor in a detergent-resistant form. Proc Natl Acad Sci USA 2008 ; 105 : 877–882. [CrossRef] [Google Scholar]
- Shibata Y, White JF, Serrano-Vega MJ, et al. Thermostabilization of the neurotensin receptor NTS1. J Mol Biol 2009 ; 390 : 262–277. [CrossRef] [PubMed] [Google Scholar]
- Magnani F, Shibata Y, Serrano-Vega MJ, Tate CG. Co-evolving stability and conformational homogeneity of the human adenosine A2a receptor. Proc Natl Acad Sci USA 2008 ; 105 : 10744–10749. [CrossRef] [Google Scholar]
- Lebon G, Bennett K, Jazayeri A, Tate CG. Thermostabilisation of an agonist-bound conformation of the human adenosine A(2A) receptor. J Mol Biol 2011 ; 409 : 298–310. [CrossRef] [PubMed] [Google Scholar]
- Warne T, Serrano-Vega MJ, Baker JG, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 2008 ; 454 : 486–491. [CrossRef] [PubMed] [Google Scholar]
- Dore AS, Robertson N, Errey JC, et al. Structure of the adenosine A(2A) receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 2011 ; 19 : 1283–1293. [CrossRef] [PubMed] [Google Scholar]
- Moukhametzianov R, Warne T, Edwards PC, et al. Two distinct conformations of helix 6 observed in antagonist-bound structures of a beta1-adrenergic receptor. Proc Natl Acad Sci USA 2011 ; 108 : 8228–8232. [CrossRef] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011 ; 469 : 175–180. [CrossRef] [PubMed] [Google Scholar]
- Steyaert J, Kobilka BK. Nanobody stabilization of G protein-coupled receptor conformational states. Curr Opin Struct Biol 2011 ; 21 : 567–572. [CrossRef] [PubMed] [Google Scholar]
- Warne T, Moukhametzianov R, Baker JG, et al. The structural basis for agonist and partial agonist action on a beta1-adrenergic receptor. Nature 2011 ; 469 : 241–244. [CrossRef] [PubMed] [Google Scholar]
- Rosenbaum DM, Zhang C, Lyons JA, et al. Structure and function of an irreversible agonist-beta2 adrenoceptor complex. Nature 2011 ; 469 : 236–240. [CrossRef] [PubMed] [Google Scholar]
- Standfuss J, Edwards PC, D’Antona A, et al. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature 2011 ; 471 : 656–660. [CrossRef] [PubMed] [Google Scholar]
- Choe HW, Kim YJ, Park JH, et al. Crystal structure of metarhodopsin II. Nature 2011 ; 471 : 651–655. [CrossRef] [PubMed] [Google Scholar]
- Scheerer P, Park JH, Hildebrand PW, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature 2008 ; 455 : 497–502. [CrossRef] [PubMed] [Google Scholar]
- Lebon G, Warne T, Edwards PC, et al. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011 ; 474 : 521–525. [CrossRef] [PubMed] [Google Scholar]
- Lebon G, Tate CG. Structure tridimensionnelle du récepteur de l’adénosine A2A lié à son agoniste naturel l’adénosine. Med Sci (Paris) 2011 ; 27 : 926–928. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Xu F, Wu H, Katritch V, et al. Structure of an agonist-bound human A2A adenosine receptor. Science 2011 ; 332 : 322–327. [CrossRef] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 2011 ; 477 : 549–555. [CrossRef] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000 ; 289 : 739–745. [CrossRef] [PubMed] [Google Scholar]
- Fritze O, Filipek S, Kuksa V, et al. Role of the conserved NPxxY(x)5, 6F motif in the rhodopsin ground state and during activation. Proc Natl Acad Sci USA 2003 ; 100 : 2290–2295. [CrossRef] [Google Scholar]
- Granier S. Structure des récepteurs mu et delta des opiacés. Med Sci (Paris) 2012 ; 28 : 870–875. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Banères JL, Mouillac B. Manipulation des RCPG : expression, purification et stabilisation in vitro. Med Sci (Paris) 2012 ; 28 : 837–844. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des tableaux
Structures des RCPG liant des ligands diffusibles. PDB : protein data bank.
Liste des figures
|
Figure 1. Illustration des différentes stratégies utilisées pour l’étude cristallographique des RCPG. A. Structure du β1-AR thermostable de la dinde (2VT4) lié à l’antagoniste cyanopindolol (vert). Les mutations thermostabilisantes sont représentées en violet. B. Structure du β2-AR-T4L complexé à l’antagoniste carazolol (jaune) (2RH1). La protéine T4L (cyan) a été fusionnée aux extrémités intracellulaires des hélices 5 et 6. C. Structure du β2-AR-T4L lié à l’agoniste BI-167107 en complexe avec un anticorps de lama nb80 (bleu marine) (3POG). D. Structure du β2-AR-T4L lié à l’agoniste BI-167107 (jaune) en complexe avec la protéine G trimérique (α en rouge, β en jaune et γ en rose) (3NS6). L’anticorps de lama nb35 (violet) stabilise la sous-unité Gαs. La protéine de fusion T4L (cyan) a été introduite à l’extrémité amino-terminale du récepteur. |
| Dans le texte | |
|
Figure 2. Conformation active R* du récepteur β 2-AR humain. A. Superposition de la structure inactive R (β2-AR lié à l’antagoniste carazolol [2HR1]) et de la conformation active R* (β2-AR complexé à l’agoniste BI-167107 et à la protéine trimérique Gs [3NS6]). Le repositionnement de l’hélice 6 ouvre une cavité dans laquelle la sous-unité Gαs s’arrime. B. Vue de la surface intracellulaire du β2-AR. Le TM5, et surtout le TM6, sont repositionnées vers l’extérieur du récepteur, alors que le TM3 et le TM7 se déplacent vers l’intérieur du récepteur. C. Vue de la surface extracellulaire du β2-AR. Le repositionnement du TM5 vers l’intérieur du récepteur s’effectue au niveau du site de fixation de l’agoniste BI-167107. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.