")
")
| Issue |
Med Sci (Paris)
Volume 27, Number 4, Avril 2011
|
|
|---|---|---|
| Page(s) | 391 - 397 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2011274015 | |
| Published online | 28 avril 2011 | |
CXCR4, une cible thérapeutique dans certains déficits immunitaires rares?
CXCR4, a therapeutic target in rare immunodeficiencies?
1 Université Paris-Sud, laboratoire cytokines, chimiokines et immunopathologie, UMR-S996, 32, rue des Carnets, 92140 Clamart, France ; Inserm, 92140 Clamart, France
2 Université Paris-Sud, laboratoire cytokines, chimiokines et immunopathologie, UMR-S996, 32, rue des Carnets, 92140 Clamart, France ; Inserm, 92140 Clamart ; AP-HP, 92140 Clamart, France
3
Université Paris Descartes, service des maladies infectieuses et tropicales, Hôpital Necker-Enfants malades, 149, rue de Sèvres, 75743 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Actuellement, plus de 200 déficits immunitaires primaires ont été identifiés. Leurs défauts génétiques affectent majoritairement l’expression ou la fonction de protéines impliquées dans le développement et la maturation du système hématopoïétique. Certaines maladies immuno-hématologiques « orphelines » sont caractérisées par un désordre de la migration leucocytaire, évocateur d’une anomalie des chimiokines et de leurs récepteurs. Cette revue dresse l’état actuel des connaissances sur les anomalies de l’axe de signalisation CXCL12 (SDF-1)/CXCR4 récemment identifiées dans la lymphopénie T CD4+ idiopathique et le syndrome WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis syndrome), deux déficits immunitaires rares chez l’homme et associés respectivement à une perte et un gain de fonction de CXCR4.
Abstract
Currently, more than 200 primary immunodeficiency diseases have been discovered. In most cases, genetic defects affect the expression or the function of proteins involved in immune development and homeostasis. Some orphan immuno-hematological disorders are characterized by an abnormal leukocyte trafficking, a notion predictive of an anomaly of the chemokine/chemokine receptor system. In this review, we focus on recent advances in the characterization of dysfunctions of the CXCL12 (SDF-1)/CXCR4 signaling axis in two rare human immunodeficiencies, one associated with a loss of CXCR4 function, the Idiopathic CD4+ T-cell Lymphocytopenia, and the other with a gain of CXCR4 function, the WHIM syndrome.
contribution égale.
© 2011 médecine/sciences – Inserm / SRMS
In memoriam
Nous dédions cette revue à la mémoire de notre collègue et directeur d’unité, le professeur
Dominique Emilie, disparu tragiquement le 25 février 2011
Les chimiokines (CK) sont de petites cytokines sécrétées et dotées d’un pouvoir chimiotactique qui gouvernent le trafic des leucocytes et le recrutement de cellules effectrices au site de l’agression. Ces activités sont relayées par des récepteurs (RCK) qui appartiennent à la famille des récepteurs à sept segments transmembranaires couplés aux protéines G, lesquels constituent les cibles de près de la moitié des médicaments disponibles [1]. Les CK/RCK exercent de multiples fonctions au sein de l’organisme, dépassant le cadre des seuls phénomènes migratoires en physio(patho)logie. Un enjeu majeur consiste à déterminer l’implication d’un, ou plusieurs, couple(s) de CK/RCK dans une pathologie d’intérêt, d’en comprendre le mode de fonctionnement et de générer des antagonistes spécifiques. Dans cette optique, la cytokine α CXC stromal cell derived factor-1 (SDF-1/CXCL12)1 et ses récepteurs constituent des cibles privilégiées pour l’élaboration de nouvelles approches pharmacologiques à visée thérapeutique.
CXCL12, une chimiokine singulière
CXCL12 est constitutivement exprimée par les cellules stromales, épithéliales et endothéliales, notamment dans les organes lymphoïdes (OL), et constitue l’unique ligand naturel de CXCR4, un RCK ubiquitaire initialement identifié comme un corécepteur du virus de l’immunodéficience humaine (VIH). CXCL12 est un facteur chimiotactique pour différentes populations leucocytaires, dont les lymphocytes T (LT), les lymphocytes B (LB) et les polynucléaires neutrophiles [2]. L’axe CXCL12/CXCR4 joue un rôle-clé dans des processus homéostatiques comme l’organogenèse, l’hématopoïèse et le trafic leucocytaire. Des dérégulations des voies de signalisation ou de l’expression de CXCR4 ont été décrites dans certaines pathologies auto-immunes (exemple : le lupus), neurodégénératives (exemple : la sclérose en plaques), cardiovasculaires (exemple : l’insuffisance coronarienne) et tumorales (exemple : le carcinome mammaire) [3]. Jusqu’en 2005, CXCL12 et CXCR4 étaient supposés maintenir une relation exclusive, compte tenu des phénotypes similaires des souris invalidées pour l’un ou l’autre gène. L’identification d’un nouveau RCK pour CXCL12, RDC1 rebaptisé CXCR7, contredit ce postulat et implique de déterminer les contributions des deux récepteurs dans les activités de CXCL12 [4].
Voies de signalisation associées à l’activation et l’inactivation de CXCR4
À la suite de la fixation de CXCL12 à CXCR4, deux phénomènes se produisent (Figure 1). Un changement conformationnel de CXCR4 déclenche l’activation des protéines G et la mobilisation de différents effecteurs intracellulaires impliqués dans la réponse cellulaire (exemple : la chimiotaxie). Ensuite, l’activation des protéines G est rapidement atténuée par un système de rétrocontrôle négatif, appelé désensibilisation, qui débute par des phosphorylations ciblant des résidus aminés de type sérine/thréonine (Ser/Thr) situés dans le domaine intracellulaire carboxy-terminal (C-ter) du RCK par des kinases spécifiques, les G protein-coupled receptor kinases (GRK). Ces réactions conduisent au recrutement de protéines adaptatrices, les β-arrestines (β-arr), qui empêchent le couplage de CXCR4 à une nouvelle protéine G et connectent le RCK à de nouvelles voies de signalisation gouvernant notamment son endocytose. Le RCK subit également des modifications post-traductionnelles (exemple : l’ubiquitinylation) qui l’orientent vers la dégradation lysosomale. Néanmoins, le RCK internalisé peut être directement recyclé à la membrane [3]. Des anomalies ciblant des étapes différentes de la signalisation de CXCR4 ont été identifiées dans deux déficits immunitaires rares chez l’homme : la lymphopénie T CD4+ idiopathique (LCI) et le syndrome WHIM (SW, ou warts, hypogammaglobulinemia, infections, and myelokathexis syndrome). La LCI est associée à une altération du recyclage membranaire de CXCR4 qui entraîne une perte de fonction du RCK, alors que le SW est lié à un défaut d’inactivation de CXCR4 qui se traduit par un gain de fonction [5, 6]. Cette revue présente comment l’étude de ces maladies a permis de décrypter le mode d’action moléculaire de l’axe CXCL12/CXCR4, et réciproquement, comment ces avancées mécanistiques ont permis de mieux comprendre leur physiopathologie et d’envisager des voies thérapeutiques futures.
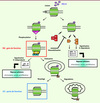 |
Figure 1 Régulation de l’activité de CXCR4. Ce schéma présente les principales voies de signalisation associées à l’activation et à l’inactivation de CXCR4 en réponse à CXCL12. Dans le SW, la désensibilisation de CXCR4 est altérée (cadre rouge), tandis que dans la LCI, il s’agit d’un défaut de recyclage membranaire de CXCR4 (cadre bleu). AMPc : adénosine monophosphate cyclique ; β-arr : β-arrestine ; ERK : extracellular signalregulated kinase ; Gαβγ : protéine G hétérotrimérique αβγ ; GRK : G protein-coupled receptor kinase ; LCI : lymphopénie T CD4+ idiopathique ; P : phosphorylation ; PI3-K : phosphoinositide 3-kinase ; PLC-β : phospholipase C-β ; SW : syndrome WHIM ; Ub : ubiquitine. |
Bases moléculaires de la perte de fonction de CXCR4 dans la lymphopénie T CD4+ idiopathique
Physiopathologie
La LCI est un déficit immunohématologique rare associant un nombre faible et persistant de LT CD4+ circulants, en l’absence d’infection par le VIH ou d’autres causes connues d’immunodéficience, et des infections opportunistes sévères [7]. Son étiologie n’impliquerait pas d’agent transmissible. Sur un plan immunologique, les LT CD4+ naïfs sont préférentiellement affectés dans le sang, alors que le nombre de LT CD8+ est préservé ou parfois diminué, constituant alors un facteur aggravant [8]. Une altération fréquente consiste en une augmentation sérique d’interleukine-7 (IL-7), compatible avec le déclenchement d’une réponse homéostatique qui vise à reconstituer le taux de LT CD4+ [9]. Des études rapportent une diminution des réponses des LT, une augmentation de leur activation et une capacité accrue à entrer en apoptose [10]. Une altération de la capacité clonogénique des cellules souches hématopoïétiques de la moelle osseuse pourrait également contribuer à la déplétion des LT CD4+ [11]. La LCI apparaît donc comme une maladie complexe, probablement multifactorielle, dont la pathogenèse reste à définir. Un enjeu consiste à déterminer si la LCI résulte d’un défaut de production thymique ou d’une consommation périphérique accélérée des LT CD4+. Les CK/RCK, et en particulier l’axe CXCL12/CXCR4, constituaient une piste à explorer. Le couple CXCL12/CXCR4 dirige la thymopoïèse et régule la domiciliation des LT dans les organes lymphoïdes secondaires [12, 13]. Une perte ou un gain d’expression de Cxcr4 chez la souris conduit à une diminution des LT CD4+ circulants associée respectivement à un défaut de la maturation thymique et à une migration exacerbée vers la moelle osseuse [14, 15]. Une altération de l’expression et/ou de l’activité membranaire de CXCR4 impacte donc sévèrement la différenciation des LT CD4+, conduisant à leur déplétion sanguine.
Association de la LCI à un défaut d’expression membranaire de CXCR4
Nous avons récemment identifié un mécanisme pathogénique impliquant CXCR4, associé à la déplétion des LT CD4+ et commun à tous les patients étudiés [5]. Les LT CD4+ circulants résiduels de 6 patients partagent un défaut d’expression membranaire de CXCR4 qui entraîne une perte de fonction, comme en témoigne l’hyposensibilité à l’action chimiotactique de CXCL12 (Figure 2). Or, l’expression et la fonction membranaires de CXCR4 dans les monocytes, et celles d’autres RCK (exemple : CCR5) dans les LT sont préservées, suggérant une anomalie restreinte aux LT et spécifique à CXCR4. Celle-ci est associée à une accumulation intracellulaire de CXCR4 et CXCL12, mais également à une incapacité de recyclage du RCK vers la surface. Aucune mutation dans CXCL12 et CXCR4 ou modification des niveaux plasmatiques de cytokines pouvant expliquer cette disparition membranaire n’a été identifiée. Sur le plan clinique, l’IL-2, in vitro et, de manière remarquable, in vivo chez certains patients, normalise l’expression membranaire de CXCR4 tout en augmentant le nombre de LT CD4+ circulants. Ainsi, cette étude ouvre pour la première fois la voie à la compréhension des anomalies moléculaires affectant les LT CD4+ et à un traitement de la LCI par immunothérapie.
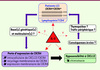 |
Figure 2 Association de la LCI à une perte de fonction de CXCR4. Ce schéma présente les principales étapes à éclaircir afin d’établir un lien causal entre l’anomalie de CXCR4 et la LCI, ainsi que les différentes hypothèses relatives à l’effet thérapeutique de l’IL-2. IL-2 : interleukine-2. |
Perte de fonction de CXCR4, une cause de la LCI ?
Ces données, qui méritent d’être étendues à d’autres cohortes, suggèrent que la perte de fonction de CXCR4 altère in vivo la différenciation des LT CD4+, conduisant à leur déplétion sanguine et favorisant ainsi l’apparition d’infections opportunistes. Plusieurs points restent à éclaircir afin d’établir un lien causal entre cette anomalie et la LCI (Figure 2). Les bases moléculaires de l’anomalie du trafic intracellulaire de CXCR4 et les conséquences in vivo sur l’homéostasie des LT CD4+ de la perte d’expression membranaire qui en résulte restent à définir. L’effet bénéfique de l’IL-2 chez certains patients suggère également une implication dans la LCI des cytokines messagères du système immunitaire dont les récepteurs partagent la chaîne γ commune (IL-2, -4, -7, -9, -15 et -21). Un défaut de réponse à ces cytokines pourrait conduire à une réduction de l’expression membranaire de CXCR4, perturbant ainsi l’homéostasie des LT CD4+. Il reste à déterminer par quel(s) mécanisme(s) moléculaire(s) l’IL-2 restaure l’expression de CXCR4 et à identifier quelle étape de la vie du LT CD4+ est normalisée par cette intervention.
Bases moléculaires du gain de fonction de CXCR4 dans le syndrome WHIM
Physiopathologie
Le SW est un déficit immunohématologique rare de transmission autosomique dominante qui se caractérise par une abondance de verrues (W, warts) liées à des infections récurrentes par le virus du papillome humain, une hypogammaglobulinémie (H), des infections bactériennes à répétition (I) et une rétention anormale de neutrophiles matures dans la moelle osseuse (M, myelokathexis) [16]. Ce tableau s’associe invariablement à une leucopénie circulante qui affecte les neutrophiles, les LT et les LB, parfois les monocytes et les cellules dendritiques [17, 18]. L’hypogammaglobulinémie, bien qu’étant variable, affecte surtout les IgG [19]. Une manifestation clinique de ces perturbations immunologiques serait la susceptibilité des patients à des épisodes infectieux itératifs, bactériens et essentiellement respiratoires [17]. Cependant, en réponse à une immunisation par un antigène T-dépendant, les patients produisent une réponse humorale spécifique, bien que les titres sériques déclinent rapidement. Le SW est donc caractérisé par un désordre de la migration leucocytaire, ce qui évoque une anomalie des CK/RCK. L’année 2003 marque un tournant décisif avec l’identification de mutations localisées dans la région codante de CXCR4 chez certains patients [20].
Association du SW à un défaut d’inactivation de CXCR4
La majorité des cas sont liés à des mutations hétérozygotes dans CXCR4 qui invalident les 10 à 19 derniers acides aminés riches en sites Ser/Thr du domaine intracellulaire carboxy-terminal du RCK, WHIMmuté(m)(Figure 3) [3, 17]. Des formes familiales sans anomalie de CXCR4 ont également été identifiées, WHIMsauvage(s), soulignant une hétérogénéité génétique du SW [6, 20]. L’intégrité du domaine carboxy-terminal étant requise pour l’inactivation de CXCR4 (Figure 1), il était logique de suspecter que les mutations altèrent ce processus et conduisent à un gain de fonction du RCK. Diverses études indiquent qu’effectivement ces différentes mutations conduisent à l’expression de RCK tronqués avec une activation accrue des protéines G, et ce malgré une expression membranaire normale [17]. Ceci s’explique par l’incapacité du CXCR4muté à être découplé des protéines G (ou désensibilisé) et internalisé en réponse à la liaison de CXCL12. Cette anomalie de l’inactivation se traduit par un gain de fonction de CXCR4, comme l’illustre l’hypersensibilité des leucocytes de ces patients à l’action chimiotactique de CXCL12. De plus, les leucocytes de patients WHIMs présentent des dysfonctionnements spécifiques de CXCR4 semblables à ceux qui sont décrits chez les patients WHIMm(Figure 3) [6]. Malgré leurs différences génétiques, les deux formes du SW présentent donc comme élément pathogénique commun un gain de fonction de CXCR4. Cependant, chez les patients WHIMm, les bases moléculaires de l’hyperactivité du RCK tronqué et son rôle étiologique dans les manifestations cliniques restaient à définir. Chez les patients WHIMs, l’activité d’autres RCK, dont CXCR7, est préservée, suggérant l’existence d’une anomalie qui affecte un régulateur de l’inactivation de CXCR4. L’étude des partenaires intracellulaires de CXCR4 a récemment permis d’apporter des éléments de réponse sur les mécanismes mis en jeu.
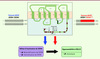 |
Figure 3 Association du SW à un défaut d’inactivation de CXCR4. Les mutations liées au SW (têtes de flèches noires dans l’encadré) affectent le domaine intracellulaire carboxy-terminal de CXCR4, lequel contient 18 sites Ser/Thr (en rouge). Pour exemple, la mutation hétérozygote ponctuelle en position 1013 (substitution S338X) conduit à l’apparition prématurée d’un codon non-sens et entraîne une délétion des 15 derniers résidus aminés, dont 10 Ser/Thr (CXCR41013). Le motif SHSK contenu dans la troisième boucle intracellulaire de CXCR4 est impliqué dans la liaison des β-arr (en vert foncé). BEC : boucle extracellulaire ; BIC : boucle intracellulaire ; C-ter : carboxy-terminal ; fs : décalage du cadre de lecture ; N-ter : amino-terminal ; S : Ser ; SHSK : Ser/His/Ser/Lys ; T : Thr ; TM : domaine transmembranaire ; WHIMm : porteurs d’une mutation dans CXCR4 ; WHIMs :sans anomalie de CXCR4. |
Altération du système GRK/β-arr dans le SW
Dans la forme WHIMm, CXCR4muté est réfractaire à la désensibilisation et à l’endocytose, ce qui signifie que l’intégrité du domaine carboxy-terminal est requise pour l’inactivation du RCK via sa phosphorylation par des GRK et la liaison des β-arr (Figure 1). En ce sens, la surexpression de β-arr ne normalise pas l’atténuation du RCK tronqué CXCR41013 (Figure 3) [21]. Cependant, CXCR41013 est capable d’interagir avec les β-arr via un domaine distinct de son extrémité carboxy-terminale : le motif Ser/His/Ser/Lys (SHSK) contenu dans la troisième boucle intracellulaire (BIC). Cette liaison est associée à l’induction de voies de signalisation G-indépendantes qui régulent positivement la chimiotaxie (Figure 1). Cet apparent paradoxe témoigne de l’ambivalence des β-arr dont les fonctions suppressives de la signalisation G-dépendante ou promotrices de la signalisation G-indépendante dépendent probablement du type de la GRK qui est recrutée et du site de fixation sur CXCR4 [3]. Ainsi, l’hypersensibilité à CXCL12 pourrait résulter d’une anomalie d’inactivation de CXCR41013 (conséquence d’un défaut de recrutement des β-arr au domaine carboxy-terminal) et d’une signalisation G-indépendante exacerbée (liée à un recrutement favorisé des β-arr à la 3e BIC). L’identification d’hétérodimères CXCR41013/CXCR4+ suggère un mécanisme par lequel CXCR4muté exerce un effet dominant négatif sur CXCR4+ [21]. Dans la forme WHIMs, nous avons dévoilé un rôle-clé de la GRK-3 dans la régulation de l’inactivation de CXCR4 [22]. L’extinction de GRK-3 dans des cellules de témoins reproduit des anomalies de CXCR4 similaires à celles documentées chez les patients. Inversement, la surexpression de GRK-3 dans les cellules de patients corrige le défaut de désensibilisation et d’internalisation de CXCR4 et normalise la chimiotaxie induite par CXCL12. Fait marquant, un déficit sélectif de la protéine GRK-3, qui résulte d’un défaut de synthèse des ARNm, a été identifié chez un des patients. Ces données révèlent une spécialisation de la GRK-3 dans l’atténuation de CXCR4 et identifient de surcroît chez les patients WHIMs une dérégulation de cette kinase comme une nouvelle anomalie liée au SW.
Interactome CXCR4/GRK/β-arr défectueux dans le SW ?
Il faut encore décrypter plusieurs étapes afin d’élucider la physiopathologie du SW et d’envisager de manipuler l’axe CXCL12/CXCR4 à des fins thérapeutiques. Dans la forme WHIMm, à l’instar de la démonstration formelle de la fonction dominante du CXCR4muté sur la forme sauvage, la spécialisation des différentes GRK reste à définir dans les voies de signalisation β-arr-dépendantes du RCK tronqué. C’est pourquoi la production de souris hétérozygotes pour une mutation dans CXCR4 est requise. La caractérisation immunologique d’un tel modèle permettra également de déterminer à quelle(s) étape(s) l’hyperactivité du CXCR4muté perturbe in vivo l’homéostasie leucocytaire et conduit à une lympho-neutropénie (Figure 4). Dans la forme WHIMs, les défauts génétiques impliqués, la nature des interactions CXCR4/GRK-3 et les conséquences in vivo d’une anomalie qui affecte la GRK-3 sur l’homéostasie leucocytaire sont à déterminer (Figure 4). La caractérisation de ces étapes est cruciale pour comprendre comment une altération de l’expression et/ou de l’activité de la GRK-3 conduit à un défaut d’inactivation de CXCR4, et par extension, aux manifestations cliniques. Son rôle pourrait impliquer la phosphorylation de sites Ser/Thr au sein du domaine carboxy-terminal de CXCR4, permettant ainsi la liaison des β-arr. Notre observation que la surexpression de GRK-3 ne normalise pas l’inactivation de CXCR41013 est un argument supplémentaire [22]. De fait, un défaut de liaison de la GRK-3 à CXCR4 pourrait contribuer au gain de fonction du RCK dans les deux formes du SW. Néanmoins, de récents travaux indiquent qu’outre la GRK-3, les GRK-2 et -6 sont capables de lier le domaine carboxy-terminal de CXCR4 à des endroits distincts et de réguler différents aspects de la signalisation [23, 24]. Cela soulève la possibilité que des défauts fonctionnels de chacune de ces kinases contribuent aux anomalies de CXCR4.
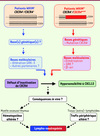 |
Figure 4 Mécanisme et impact sur l’homéostasie leucocytaire du gain de fonction de CXCR4 dans le SW. Ce schéma rapporte les avancées récentes dans la compréhension des bases moléculaires du gain de fonction de CXCR4 et indique les étapes-clés à caractériser afin d’élucider la physiopathologie du SW. |
Conclusion
La LCI et le SW possèdent un dénominateur commun, une anomalie de CXCR4, qui affecte cependant des étapes différentes de la signalisation du RCK, ce qui conduit à des phénotypes fonctionnels opposés. La LCI est associée à un défaut de recyclage membranaire de CXCR4 qui entraîne une perte de fonction, tandis que le SW est lié à un défaut d’inactivation du RCK qui se traduit par un gain de fonction. Ces anomalies pourraient affecter des stades distincts de la vie des leucocytes, ce qui aurait pour conséquence une déplétion des LT CD4+ dans la LCI et une leucopénie dans le SW, mais ceci doit être élucidé. Un modèle animal du SW serait d’un apport indéniable, d’autant qu’il permettrait d’évaluer les vertus pharmacologiques des inhibiteurs de l’axe CXCL12/CXCR4. L’AMD3100, un antagoniste sélectif de CXCR4 et connu pour ses propriétés mobilisatrices, constitue un bon candidat et pourrait orienter l’arsenal thérapeutique chez les patients dont la prise en charge n’est actuellement pas spécifique [25]. Dans la LCI, il reste à déterminer le rôle étiologique de l’anomalie de CXCR4, d’autant que l’hétérogénéité des profils immunologiques des patients suggère une origine multifactorielle. L’étude princeps montre qu’un traitement in vitro ou in vivo par l’IL-2 normalise la fonction de CXCR4. Le seul patient chez lequel aucune amélioration du taux de LT CD4+ n’est observée lors de ce traitement est celui pour lequel l’anomalie de CXCR4 est réfractaire aux effets bénéfiques de l’IL-2 [5]. Ceci soulève la possibilité qu’une correction de l’anomalie de CXCR4 in vitro soit prédictive d’un succès thérapeutique. La modélisation de ces maladies permettra également d’identifier de nouveaux partenaires moléculaires de CXCR4 et de définir leur contribution à l’activité du RCK. Ces candidats pourraient constituer les cibles de nouvelles approches thérapeutiques. Enfin, on peut concevoir d’instaurer une veille systématique des immunodéficiences « non étiquetées » dont la sémiologie suggère un désordre de la migration leucocytaire, et de fait, une anomalie des RCK. L’identification d’un nouveau déficit immunitaire lié à une anomalie de CXCR4, différent de la LCI et du SW et prédisposant à des infections à mycobactéries, en est le parfait exemple [26].
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Remerciements
Les auteurs souhaitent remercier l’Institut national de la santé et de la recherche médicale (Inserm), l’Assistance publique-Hôpitaux de Paris (AP-HP, contrat n° 07018), l’Université Paris-Sud, l’Union européenne (FP6, INNOCHEM, contrat n° LSHB-CT-2005-518167), l’Agence nationale de la recherche (ANR, contrat Jeunes chercheurs n° 2010JCJC110401), l’Association pour la recherche sur le cancer (ARC, contrat n° 4982) et la Ligue nationale contre le cancer (comité Val d’Oise) pour leur soutien financier.
Les chimiokines sont classées en quatre groupes selon le nombre d’acides aminés présents entre les deux premières cystéines situées dans la partie amino-terminale : CC, CXC, CX3C ainsi que le groupe C qui comprend la lymphotactine (XCL1). Le groupe CXC est encore subdivisé en deux catégories en fonction de la présence ou de l’absence d’un motif ELR (glutamate-leucine-arginine) en amont du premier résidu cystéine. Cette classification structurale fait aujourd’hui office de nomenclature de référence mais les noms communs donnés précédemment aux chimiokines sont encore largement utilisés, comme par exemple SDF-1 (CXCL12), SLC (CCL21), ELC (CCL19), MIP-3a (CCL20), etc.
Références
- MackayCR. Moving targets: cell migration inhibitors as new anti-inflammatory therapies. Nat Immunol 2008 ; 9 : 988-998. [CrossRef] [PubMed] [Google Scholar]
- DesjardinsSF, BerchicheYA, HaddadE, HevekerN.. CXCR4, un récepteur de chimiokine aux multiples talents. Med Sci (Paris) 2007 ; 23 : 980-984. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- BusilloJM, BenovicJL. Regulation of CXCR4 signaling. Biochim Biophys Acta 2007 ; 1768 : 952-963. [CrossRef] [PubMed] [Google Scholar]
- BalabanianK, LaganeB, InfantinoS, et al. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem 2005 ; 280 : 35760-35766. [CrossRef] [PubMed] [Google Scholar]
- Scott-AlgaraD, BalabanianK, ChakrabartiLA, et al. Idiopathic CD4+ T-cell lymphocytopenia is associated with impaired membrane expression of the chemokine receptor CXCR4. Blood 2010 ; 115 : 3708-3717. [CrossRef] [PubMed] [Google Scholar]
- BalabanianK, LaganeB, PablosJL, et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood 2005 ; 105 : 2449-2457. [CrossRef] [PubMed] [Google Scholar]
- LuoL, LiT. Idiopathic CD4 lymphocytopenia and opportunistic infection-an update. FEMS Immunol Med Microbiol 2008 ; 54 : 283-289. [CrossRef] [PubMed] [Google Scholar]
- ZoniosDI, FalloonJ, BennettJE, et al. Idiopathic CD4+ lymphocytopenia: natural history and prognostic factors. Blood 2008 ; 112 : 287-294. [CrossRef] [PubMed] [Google Scholar]
- MalaspinaA, MoirS, ChaittDG, et al. Idiopathic CD4+ T lymphocytopenia is associated with increases in immature/transitional B cells and serum levels of IL-7. Blood 2007 ; 109 : 2086-2088. [CrossRef] [PubMed] [Google Scholar]
- WalkerUA, WarnatzK. Idiopathic CD4 lymphocytopenia. Curr Opin Rheumatol 2006 ; 18 : 389-395. [CrossRef] [PubMed] [Google Scholar]
- IsgroA, SirianniMC, GramiccioniC, et al. Idiopathic CD4+ lymphocytopenia may be due to decreased bone marrow clonogenic capability. Int Arch Allergy Immunol 2005 ; 136 : 379-384. [CrossRef] [PubMed] [Google Scholar]
- Cavazzana-CalvoM, SixE, Andre-SchmutzI, CoulombelL. Hématopoïèse humaine : des cellules CD34 aux lymphocytes T. Med Sci (Paris) 2007 ; 23 : 151-159. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- BromleySK, MempelTR, LusterADOrchestrating the orchestrators: chemokines in control of T cell traffic. Nat Immunol 2008 ; 9 : 970-980. [CrossRef] [PubMed] [Google Scholar]
- OnaiN, ZhangY, YoneyamaH, et al. Impairment of lymphopoiesis and myelopoiesis in mice reconstituted with bone marrow-hematopoietic progenitor cells expressing SDF-1-intrakine. Blood 2000 ; 96 : 2074-2080. [PubMed] [Google Scholar]
- SawadaS, GowrishankarK, KitamuraR, et al. Disturbed CD4+ T cell homeostasis and in vitro HIV-1 susceptibility in transgenic mice expressing T cell line-tropic HIV-1 receptors. J Exp Med 1998 ; 187 : 1439-1449. [CrossRef] [PubMed] [Google Scholar]
- GorlinRJ, GelbB, DiazGA, et al. WHIM syndrome, an autosomal dominant disorder: clinical, hematological, and molecular studies. Am J Med Genet 2000 ; 91 : 368-376. [CrossRef] [PubMed] [Google Scholar]
- KawaiT, MalechHL. WHIM syndrome: congenital immune deficiency disease. Curr Opin Hematol 2009 ; 16 : 20-26. [CrossRef] [PubMed] [Google Scholar]
- TassoneL, MorattoD, VermiW, et al. Defect of plasmacytoid dendritic cells in warts, hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome patients. Blood 2010 ; 116 : 4870-4873. [CrossRef] [PubMed] [Google Scholar]
- Mc GuirePJ, Cunningham-RundlesC, OchsH, DiazGA.. Oligoclonality, impaired class switch and B-cell memory responses in WHIM syndrome. Clin Immunol 2010 ; 135 : 412-421. [CrossRef] [PubMed] [Google Scholar]
- HernandezPA, GorlinRJ, LukensJN, et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet 2003 ; 34 : 70-74. [CrossRef] [PubMed] [Google Scholar]
- LaganeB, ChowKY, BalabanianK, et al. CXCR4 dimerization and beta-arrestin-mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood 2008 ; 112 : 34-44. [CrossRef] [PubMed] [Google Scholar]
- BalabanianK, LevoyeA, KlemmL, et al. Leukocyte analysis from WHIM syndrome patients reveals a pivotal role for GRK3 in CXCR4 signaling. J Clin Invest 2008 ; 118 : 1074-1084. [PubMed] [Google Scholar]
- McCormickPJ, SegarraM, GasperiniP, et al. Impaired recruitment of Grk6 and beta-Arrestin 2 causes delayed internalization and desensitization of a WHIM syndrome-associated CXCR4 mutant receptor. PLoS One 2009 ; 4 : e8102. [Google Scholar]
- BusilloJM, ArmandoS, SenguptaR, et al. Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J Biol Chem 2010 ; 285 : 7805-7817. [CrossRef] [PubMed] [Google Scholar]
- PusicI, DiPersioJF. Update on clinical experience with AMD3100, an SDF-1/CXCL12-CXCR4 inhibitor, in mobilization of hematopoietic stem and progenitor cells. Curr Opin Hematol 2010 ; 17 : 319-326. [CrossRef] [PubMed] [Google Scholar]
- DonckerAV, BalabanianK, Bellanne-ChantelotC, et al. Two cases of disseminated Mycobacterium avium infection associated with a new immunodeficiency syndrome related to CXCR4 dysfunctions. Clin Microbiol Infect 2011 ; 17 : 135-139. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1 Régulation de l’activité de CXCR4. Ce schéma présente les principales voies de signalisation associées à l’activation et à l’inactivation de CXCR4 en réponse à CXCL12. Dans le SW, la désensibilisation de CXCR4 est altérée (cadre rouge), tandis que dans la LCI, il s’agit d’un défaut de recyclage membranaire de CXCR4 (cadre bleu). AMPc : adénosine monophosphate cyclique ; β-arr : β-arrestine ; ERK : extracellular signalregulated kinase ; Gαβγ : protéine G hétérotrimérique αβγ ; GRK : G protein-coupled receptor kinase ; LCI : lymphopénie T CD4+ idiopathique ; P : phosphorylation ; PI3-K : phosphoinositide 3-kinase ; PLC-β : phospholipase C-β ; SW : syndrome WHIM ; Ub : ubiquitine. |
| Dans le texte | |
|
Figure 2 Association de la LCI à une perte de fonction de CXCR4. Ce schéma présente les principales étapes à éclaircir afin d’établir un lien causal entre l’anomalie de CXCR4 et la LCI, ainsi que les différentes hypothèses relatives à l’effet thérapeutique de l’IL-2. IL-2 : interleukine-2. |
| Dans le texte | |
|
Figure 3 Association du SW à un défaut d’inactivation de CXCR4. Les mutations liées au SW (têtes de flèches noires dans l’encadré) affectent le domaine intracellulaire carboxy-terminal de CXCR4, lequel contient 18 sites Ser/Thr (en rouge). Pour exemple, la mutation hétérozygote ponctuelle en position 1013 (substitution S338X) conduit à l’apparition prématurée d’un codon non-sens et entraîne une délétion des 15 derniers résidus aminés, dont 10 Ser/Thr (CXCR41013). Le motif SHSK contenu dans la troisième boucle intracellulaire de CXCR4 est impliqué dans la liaison des β-arr (en vert foncé). BEC : boucle extracellulaire ; BIC : boucle intracellulaire ; C-ter : carboxy-terminal ; fs : décalage du cadre de lecture ; N-ter : amino-terminal ; S : Ser ; SHSK : Ser/His/Ser/Lys ; T : Thr ; TM : domaine transmembranaire ; WHIMm : porteurs d’une mutation dans CXCR4 ; WHIMs :sans anomalie de CXCR4. |
| Dans le texte | |
|
Figure 4 Mécanisme et impact sur l’homéostasie leucocytaire du gain de fonction de CXCR4 dans le SW. Ce schéma rapporte les avancées récentes dans la compréhension des bases moléculaires du gain de fonction de CXCR4 et indique les étapes-clés à caractériser afin d’élucider la physiopathologie du SW. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.