")
")
| Issue |
Med Sci (Paris)
Volume 25, Number 2, Février 2009
|
|
|---|---|---|
| Page(s) | 175 - 180 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2009252175 | |
| Published online | 15 février 2009 | |
Guider et intégrer pour un épissage diversifié
Guiding and integrating to control and diversify splicing
Chaire de recherche du Canada en génomique fonctionnelle, Groupe ARN, Département de microbiologie et d’infectiologie, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, 3001, 12 e Avenue Nord, Sherbrooke, Québec J1H 5N4, Canada
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Trente années se sont écoulées depuis la découverte de l’épissage alternatif et notre compréhension des mécanismes de régulation demeure encore très incomplète malgré l’importance de ce processus dans les maladies humaines. Des percées récentes ont néanmoins permis d’identifier une panoplie d’activateurs et de répresseurs liant des séquences introniques et exoniques. L’expression différentielle de ces facteurs et leur utilisation combinée permettraient à la cellule d’effectuer le choix des sites d’épissage de façon précise et spécifique. Afin de conjuguer ces décisions avec d’autres processus cellulaires, certains de ces régulateurs sont recrutés durant la transcription, et leur activité est souvent intégrée aux voies de signalisation. Des efforts soutenus combinant approches classiques et technologies de pointe devraient confirmer le rôle exceptionnel de l’épissage alternatif comme agent diversificateur du protéome et améliorer notre connaissance des réseaux de régulation sous-jacents.
Abstract
Recent studies directed at understanding alternative splicing control have produced an expanding list of regulators that can enhance or silence the use of splice sites by binding to specific sequences. A fine balance in the expression and the combinatorial use of these factors would help to adapt splicing decisions to a variety of situations. Additional levels of control are provided by tightly connecting the activity of alternative splicing factors with other cellular processes such as signal transduction and transcription. Combining classical experiments and high-throughput approaches is now confirming the important contribution of alternative splicing to proteomic diversity while helping to decipher the underlying networks of splicing regulation.
© 2009 médecine/sciences - Inserm / SRMS
Le séquençage du génome humain a révélé que le fonctionnement biologique de l’organisme le plus complexe de la planète ne reposait que sur 25 000 gènes seulement, un nombre légèrement plus élevé que celui exprimé chez le ver Caenorhabditis elegans (environ 20 000) et considérablement moins grand que celui du riz (environ 35 000). Comme le nombre de gènes n’est de toute évidence pas un bon indicateur de la complexité biologique, d’autres perspectives moléculaires doivent être envisagées. L’utilisation de combinaisons précises de gènes associée à la régulation temporelle de leur expression pourrait justifier cette complexité qui caractérise les organismes supérieurs. De plus, un mécanisme biologique très important nommé épissage alternatif conduirait à un niveau supérieur de diversité et de fonctionnalité des gènes.
La très grande majorité des gènes humains est constituée d’exons interrompus par des introns. Pour chacun des gènes transcrits en ARN pré-messager (pré-ARNm), l’épissage des introns permet de produire un ARN messager (ARNm) constitué d’exons dès lors positionnés de façon consécutive. L’épissage alternatif est le processus par lequel certains exons, certains introns ou des portions de ceux-ci, sont alternativement gardés ou enlevés (Figure 1). Comme plus de 70 % des gènes humains sont épissés de façon alternative [1] et que le nombre d’isoformes produits à partir d’un seul gène peut être important [2], ce processus permet à un organisme de produire un protéome beaucoup plus complexe que celui émanant d’un réservoir limité de gènes toujours épissés de façon uniforme.
 |
Figure 1. Les modes d’épissage alternatif. Les modes d’épissage alternatif des pré-ARNm incluent l’exclusion d’exons, l’inclusion mutuellement exclusive d’exons, la concurrence entre sites d’épissage 5’, la concurrence entre sites d’épissage 3’ et la rétention d’introns. |
Bien que les cellules puissent produire une variété importante d’ARNm, des dérèglements dans les niveaux relatifs d’isoformes peuvent causer certaines maladies graves. Dans certaines situations, des mutations ciblant directement des séquences régulatrices peuvent modifier la sélection des sites d’épissage alternatif. Par exemple, des maladies telles que la déficience familiale isolée en hormone de croissance type II (IGHD II), la démence fronto-temporale avec parkinsonisme liée au chromosome 17 (FTDP-17) et la mucoviscidose sont causées par ce type d’altération [3]. De plus, un changement dans les niveaux d’expression de certains facteurs d’épissage alternatif peut influencer l’apparition de certains cancers [4, 5].
Les règles de l’épissage
Un épissage réussi repose sur la capacité d’une machinerie cellulaire à lier chacun des exons de façon précise et efficace. La première étape du processus est assez bien comprise : l’une des extrémités de l’intron, soit le site d’épissage 5’, est reconnue par un complexe ribonucléoprotéique appelé snRNP (small nuclear ribonucleoprotein) U1. La reconnaissance de l’autre extrémité, le site d’épissage 3’ constitué d’un site de branchement, d’une séquence riche en pyrimidines et du dinucléotide AG, exige la participation de plusieurs facteurs dont le snRNP U2 et l’hétérodimère U2AF65/U2AF35 (Figure 2A). L’efficacité avec laquelle les extrémités des exons sont reconnues est établie le plus fréquemment à partir des séquences qu’on y retrouve. Lorsque ces sites de liaison ne sont pas optimaux, un snRNP U1 lié à un site d’épissage 5’ pourra aider à mieux discerner un site d’épissage 3’ en amont, contribuant ainsi à définir l’exon (Figure 2A). Dans d’autres situations, des facteurs spécifiques comme les protéines SR peuvent lier des séquences situées à proximité des sites d’épissage pour favoriser leur reconnaissance par la machinerie splicéosomale (Figure 2B).
 |
Figure 2. La reconnaissance des signaux d’épissage. A. La reconnaissance du site d’épissage 5’ (5’ss) (A/CAGGUA/GAGU) s’effectue via le snRNP U1, alors que la reconnaissance du site de branchement de la région riche en pyrimidines et du dinucléotide AG met en jeu respectivement le snRNP U2, U2AF65 et U2AF35. La reconnaissance du site d’épissage 3’ (3’ss) peut être couplée à la liaison du U1 snRNP en aval, contribuant ainsi à la définition de l’exon. B. Des séquences exoniques liées par les protéines SR permettent d’augmenter la reconnaissance des signaux d’épissage par le splicéosome. C. L’interaction entre protéines hnRNP liées près de sites d’épissage distants permet un rapprochement des extrémités de l’intron et contribue à inhiber l’utilisation de pseudo sites introniques. D. La liaison de SRp30c et ASF/SF2 à des régions introniques peut inhiber l’utilisation de sites d’épissage 3’. E. La liaison du régulateur PTB à une séquence intronique contribue à inactiver l’assemblage du spliceosome en inhibant l’interaction entre les snRNP U1 et U2. Lorsqu’il est lié dans l’exon, PTB peut inactiver la définition de l’exon en inhibant le couplage entre U2AF et le snRNP U1. La liaison de la protéine hnRNP A1 à une région exonique peut nuire à l’utilisation du site d’épissage 3’ situé en amont. F. L’expression d’un régulateur pourrait avoir des effets opposés dans différents types cellulaires. Dans l’exemple montré, un effet positif pourrait être attribué au facteur X dans l’épissage d’un exon alternatif pour le type cellulaire A. Cependant, dans le type cellulaire B, l’expression de la protéine X pourrait interférer avec la liaison d’un facteur plus puissant (Y). Le déplacement de Y par X dans le type cellulaire B diminuerait ainsi l’inclusion de l’exon alternatif. |
Un défi actuel considérable consiste à comprendre comment le complexe au site d’épissage 5’ trouve celui qui est assemblé au site d’épissage 3’ en aval. En effet, comme la taille des introns humains peut atteindre plusieurs milliers de nucléotides [6], on peut être intrigué par les mécanismes qui permettent de jumeler les jonctions d’épissage authentiques tout en évitant l’utilisation de pseudo-sites que l’on retrouve immanquablement dans les grands introns. Un mécanisme proposé par notre équipe met en cause la protéine hnRNP A1 liée à des séquences introniques près des jonctions d’épissage [7]. Une interaction entre protéines A1 liées favoriserait le rapprochement de complexes d’épissage distants tout en inhibant les sites d’épissage situés entre les séquences de liaison pour A1 (Figure 2C). Ce type d’interactions ne serait pas limité à hnRNP A1 car hnRNP H [8] et Nova2 [9] pourraient également promouvoir des changements dans la conformation du pré-ARNm afin de contribuer à identifier les partenaires d’épissage. Une fois les extrémités de l’intron jumelées, l’assemblage complet du splicéosome par l’ajout de snRNP et d’autres protéines aboutirait aux deux réactions successives de trans-estérification menant à la ligation des deux exons et à l’excision de l’intron [10].
Bien que l’utilisation des sites d’épissage alternatifs soit soumise aux mêmes règles énoncées plus haut pour l’épissage générique, au moins deux propriétés supplémentaires contribuent à empêcher que ces sites d’épissage ne soient utilisés de façon systématique. Premièrement, leurs séquences n’adhèrent que faiblement aux consensus définissant des sites d’épissage efficaces. Deuxièmement, des régions introniques et/ou exoniques vont souvent mener au recrutement de facteurs qui inhiberont l’utilisation de ces sites. Ainsi, la liaison de protéines SR à certaines régions introniques peut avoir un effet négatif. C’est le cas pour ASF/SF2 et SRp30c qui inhibent la disponibilité de certains sites d’épissage 3’ alternatifs [11, 12] (Figure 2D). D’autres protéines comme hnRNP A1 et PTB répriment aussi l’usage de certains sites d’épissage, soit en empêchant leur reconnaissance [13], soit en obstruant la communication entre ces sites [14, 15] (Figure 2E). Néanmoins, la liaison de certaines protéines activatrices peut contrer ces barrages et ainsi favoriser l’épissage au niveau de certains sites.
La complexité : le prix à payer pour être spécifique
Beaucoup d’événements d’épissage alternatif se produisent plus fréquemment dans un tissu ou lors d’une situation biologique particulière [16]. Il est possible qu’un facteur exprimé de façon histo-spécifique puisse induire plusieurs décisions d’épissage : c’est le cas des régulateurs neuro-spécifiques Nova2 et nPTB [9, 17]. L’exemple de l’exon N1 du gène c-src illustre bien la contribution de ce type de régulation. Parmi les huit protéines participant à l’orchestration de cet épissage alternatif, la protéine PTB entraîne l’exclusion de l’exon N1 dans les cellules non neuronales, tandis que dans les cellules neuronales, l’homologue neuro-spécifique nPTB empêche la liaison de PTB, favorisant ainsi l’inclusion de l’exon N1 [18]. Outre Nova2 et nPTB, il existe pour le moment peu d’exemples de protéines régulatrices exprimées de façon clairement histo-spécifique.
Comme le nombre de gènes épissés de façon alternative est élevé, chaque unité ne peut avoir son propre régulateur. Une façon plus répandue d’influencer la sélection d’un site d’épissage alternatif consiste à mettre en jeu plusieurs protéines régulatrices ; la décision finale étant le résultat de la contribution de tous ces facteurs. L’expression de ces protéines peut différer d’un type cellulaire à un autre [19] et des changements dans la concentration cellulaire de ces régulateurs provoquent des profils d’épissage alternatif de façon spécifique [20]. Les niveaux relatifs de transcription des gènes encodant ces régulateurs seraient donc très importants. De plus, l’expression de ces facteurs peut être sous le contrôle de micro-ARN. Par exemple, lors de la différenciation neuronale, miR-124 diminue les niveaux de PTB, contribuant ainsi à une transition neuro-spécifique du profil d’épissage alternatif de certains gènes [21]. Lors de la maturation musculaire, c’est l’expression du facteur nPTB qui est soumis à l’action de miR-133, altérant ainsi le profil d’épissage alternatif de gènes mobilisés dans la différenciation des myoblastes [22].
Dans les cas où des différences d’expression semblent absentes, un autre moyen de moduler la sélection des sites d’épissage aurait pour point de départ une modification de l’activité de ces régulateurs ou de leur localisation cellulaire en réponse à des signaux intra- et/ou extracellulaires. Des différences mineures dans ces niveaux pourraient avoir des effets importants si ces protéines coopèrent normalement avec d’autres facteurs pour produire un effet synergétique dans la régulation.
La spécificité d’action est aussi imposée par la position relative du site de liaison du facteur sur l’unité d’épissage alternatif. Par exemple, la liaison de hnRNP H à proximité du site d’épissage 3’ peut être répressive [23], tandis qu’elle stimule l’épissage lorsqu’elle est localisée dans l’intron de gènes comme c-src et Bcl-x [24, 25]. De même, la liaison de la protéine Fox2 en amont de certains exons provoque leur exclusion [26], tandis que l’inclusion de plusieurs exons est stimulée lorsque Fox2 est liée en aval [27]. Sur le pré-ARNm du récepteur R1 du NMDA, la protéine NAPOR (neuroblastoma apoptosis-related RNA-binding protein) favorise l’exclusion de l’exon 5, et l’inclusion de l’exon 21 sur le même pré-ARNm [28]. Ces situations illustrent l’importance du contexte sur le mode d’action d’une protéine régulatrice. On peut imaginer qu’un régulateur pourrait même avoir des effets opposés sur un événement d’épissage donné s’il interfère avec l’activité d’un facteur plus puissant exprimé seulement dans un type cellulaire (Figure 2F).
Transcrire et épisser
La transcription peut influencer l’épissage alternatif de quatre façons distinctes. Premièrement, comme la force d’un promoteur détermine la quantité des transcrits produits, une stimulation dans l’expression d’un gène pourrait rendre insuffisant le niveau d’un régulateur entraînant un changement dans la sélection des sites d’épissage (Figure 3A). Deuxièmement, l’assemblage de complexes transcriptionnels distincts à un promoteur pourrait changer la localisation nucléaire de l’unité de transcription pour une région plus richement pourvue en facteurs de régulation spécifiques (Figure 3B). Troisièmement, un régulateur d’épissage pourrait être recruté par un complexe transcriptionnel précis ou par l’intermédiaire du domaine carboxy-terminal (CTD) de l’ARN polymérase II. Par exemple, l’inhibition de l’inclusion d’un exon alternatif de la fibronectine dépendrait directement du recrutement de SRp20 par le domaine CTD de l’ARN polymérase II [29]. L’interaction des facteurs d’épissage avec ce domaine étant modulée par l’état de phosphorylation du CTD [30], cette capacité serait très variable puisque le patron de phosphorylation du CTD change au cours de la transcription [31]. Il est donc possible d’envisager que des modifications ciblant le CTD et certains facteurs d’épissage moduleraient leurs interactions au cours d’une ronde transcriptionnelle avec des conséquences sur les profils d’épissage alternatif. Finalement, un couplage entre la transcription et l’épissage peut aussi reposer sur la vitesse d’élongation de l’ARN polymérase II. Un ralentissement de cette vitesse procurerait plus de temps à la machinerie d’épissage pour un assemblage sur des sites faibles [32]. Des sites de pause internes pour la polymérase auraient un effet similaire à condition que ceux-ci soient positionnés adéquatement [33].
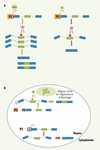 |
Figure 3. Couplage entre la transcription et l’épissage. A. Un gène sous le contrôle d’un promoteur (rectangle orange, P1) produit une grande quantité d’ARN pré-messager dans un premier cas (gauche) et, dans d’autres conditions, ce gène exprime une moins grande quantité de transcrits (droite). Une protéine régulatrice (cercle rouge) ayant une activité négative sur un exon alternatif (rectangle vert) de cet ARN pré-messager sera en quantité insuffisante pour contrôler tous les exons alternatifs dans le cas où le transcrit est produit en trop grande quantité. Les rectangles bleus sont des exons constitutifs.B. Tout dépendant lequel des promoteurs est utilisé (rectangle orange ou rectangle mauve), le pré-ARN messager produit pourrait être transporté dans des régions différentes du noyau. Par exemple, lorsque le gène est exprimé sous le contrôle du promoteur 1 (P1, rectangle orange) (partie supérieure), le transcrit est envoyé dans une région riche en protéines régulatrices (cercles verts) et l’exon alternatif (rectangle vert) sera modulé par ces facteurs. Les rectangles bleus sont des exons constitutifs. |
Le couplage entre la transcription et l’épissage alternatif s’inscrit fort probablement dans un processus dynamique plus global où entre en jeu la structure de la chromatine. Ainsi, le complexe de remodelage de la chromatine SWI/SNF diminuerait la vitesse d’élongation de l’ARN polymérase II et provoquerait l’accumulation de l’enzyme à un endroit précis d’une unité d’épissage alternatif [34]. De plus, certains composants de la chromatine pourraient médier les interactions entre le CTD et certaines kinases. À la suite d’une première ronde transcriptionnelle, une série de modifications de la chromatine pourrait accélérer la vitesse de transcription des rondes successives [35], avec des conséquences sur les profils d’épissage alternatif de ce gène.
Des signaux à intégrer
La plupart des régulateurs d’épissage subissent des modifications post-traductionnelles telles la phosphorylation. Ainsi plusieurs kinases dont SRPK (SR proteinspecific kinase-1), CLK/STY, PRP4K, CDK11 (cyclin-dependent kinase 11)/PITSLRE et CDK12/CrkRS (Cdc2-related kinase), et au moins une phosphatase (PP1) jouent un rôle dans la modulation de l’activité des protéines SR. Bien que l’on ignore encore l’étendue des événements de signalisation qui influencent l’épissage alternatif, certaines voies incluant Ras/MEK/ERK, Rac/JNK/p38 MAPK, Ras/PI3K/Akt et Ca2+/calmoduline/CaMKIV ont répertoriées [36]. Parmi les exemples les mieux étudiés, la voie Ras/Raf/MEK/ERK régule étroitement l’épissage alternatif de l’exon variable 5 (v5) de CD44. La protéine de liaison à l’ARN Sam68 a également été reconnue comme une cible directe pour ERK, sa phosphorylation stimulant l’inclusion de l’exon v5 de CD44 [37].
L’étude détaillée des mécanismes de régulation commence à révéler l’existence de voies convergentes assurant une quantité précise des variants d’épissage. Pour le gène apoptotique Bcl-x, par exemple, plusieurs protéines régulatrices et voies de signalisation contribuent à définir la production de deux isoformes : la version anti-apoptotique Bcl-xL et la forme Bcl-xS qui encourage, au contraire, la mort cellulaire [38]. Parmi les protéines régulatrices connues, on compte certaines protéines SR comme SRp30c, des protéines hnRNP comme A1, F et H et d’autres facteurs comme Sam68 (68-kDa Src-associated protein) et SAP155 [25, 39–41] (Figure 4).
 |
Figure 4. Protéines régulatrices participant à l’épissage alternatif de Bcl-x. L’épissage alternatif du gène Bcl-x engendre une isoforme pro-apoptotique ou anti-apoptotique. La voie de signalisation de la protéine kinase C (PKC) est en cause dans l’inhibition du site d’épissage par l’intermédiaire d’un facteur encore inconnu. La protéine SAP155 inhibe l’utilisation du site d’épissage xS, alors que Sam68, hnRNP A1, F et H favorisent la production de l’isoforme Bcl-xS. SRp30c stimule l’utilisation du site d’épissage 5’ de Bcl-xL. |
Étant donné les conséquences biologiques dramatiques de l’épissage sur Bcl-x, il n’est pas étonnant que ce processus soit étroitement intégré aux voies de signalisation qui surveillent l’état de santé de la cellule et sa relation avec son environnement. En effet, l’épissage alternatif menant à la synthèse de Bcl-xS est encouragé par certains stress qui inactivent PP1 [41]. Alternativement, des cytokines telles que IL(interleukine)-6 et GM-CSF (granulocyte-macrophage colony-stimulating factor) augmentent la production de l’isoforme Bcl-xL [42]. D’autre part, la kinase Fyn inhibe l’activité de Sam68 ce qui augmente la production de Bcl-xL [40]. Finalement, bien que PKC puisse réprimer l’utilisation de Bcl-xS dans certaines cellules, les cellules cancéreuses semblent utiliser des voies de régulation alternatives qui pourraient assurer une répression plus forte visant à promouvoir une meilleure résistance à l’apoptose [43].
Conclusions
La bio-informatique associée au développement de tests automatisés et à la récolte de données, par l’analyse de micropuces et le séquençage de masse, permet de prévoir une évolution rapide de notre compréhension des stratégies et des réseaux de régulation au cœur des décisions d’épissage dans différents types cellulaires. Comme ces décisions sont souvent altérées dans certaines pathologies comme le cancer, l’application des connaissances qui expliquent comment les différences d’expression, les mutations et les polymorphismes entre individus contribuent au développement des maladies et à leur évolution chez l’homme représente un enjeu important au plan médical. Force est de constater que l’incroyable diversité créée par l’épissage alternatif contribue actuellement à l’identification de nouveaux biomarqueurs à potentiel clinique [44]. Il est maintenant permis d’espérer qu’une meilleure caractérisation des isoformes d’épissage et des mécanismes régulant leur production mènera à des traitements innovateurs en vue d’améliorer la santé humaine.
Références
- Johnson JM, Castle J, Garrett-Engele P, et al. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 2003; 302 : 2141–4. [Google Scholar]
- Lipscombe D. Neuronal proteins custom designed by alternative splicing. Curr Opin Neurobiol 2005; 15 : 358–63. [Google Scholar]
- Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev 2003; 17 : 419–37. [Google Scholar]
- Karni R, de Stanchina E, Lowe SW, et al. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol 2007; 14 : 185–93. [Google Scholar]
- Ghigna C, Giordano S, Shen H, et al. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell 2005; 20 : 881–90. [Google Scholar]
- Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 2003; 72 : 291–336. [Google Scholar]
- Martinez-Contreras R, Cloutier P, Shkreta L, et al. hnRNP proteins and splicing control. Adv Exp Med Biol 2007; 623 : 123–47. [Google Scholar]
- Martinez-Contreras R, Fisette JF, Nasim FU, et al. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol 2006; 4 : e21. [Google Scholar]
- Ule J, Stefani G, Mele, et al. An RNA map predicting Nova-dependent splicing regulation. Nature 2006; 444 : 580–6. [Google Scholar]
- McGrail JC, Tatum EM, O’Keefe RT. Mutation in the U2 snRNA influences exon interactions of U5 snRNA loop 1 during pre-mRNA splicing. EMBO J 2006; 25 : 3813–22. [Google Scholar]
- Simard MJ, Chabot B. SRp30c is a repressor of 3’ splice site utilization. Mol Cell Biol 2002; 22 : 4001–10. [Google Scholar]
- Kanopka A, Muhlemann O, Akusjarvi G. Inhibition by SR proteins of splicing of a regulated adenovirus pre-mRNA. Nature 1996; 381 : 535–8. [Google Scholar]
- Domsic JK, Wang Y, Mayeda A, et al. Human immunodeficiency virus type 1 hnRNP A/B-dependent exonic splicing silencer ESSV antagonizes binding of U2AF65 to viral polypyrimidine tracts. Mol Cell Biol 2003 23 : 8762–72. [Google Scholar]
- Izquierdo JM, Majós N, Bonnal S, et al. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol Cell 2005; 19 : 475–84. [Google Scholar]
- Sharma S, Kohlstaedt LA, Damianov A, et al. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat Struct Mol Biol 2008; 15 : 183–91. [Google Scholar]
- Blencowe BJ. Alternative splicing : new insights from global analyses. Cell 2006; 126 : 37–47. [Google Scholar]
- Boutz PL, Stoilov P, Li Q, et al. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev 2007; 21 : 1636–52. [Google Scholar]
- Markovtsov V, Nikolic JM, Goldman JA, et al. Cooperative assembly of an hnRNP complex induced by a tissue-specific homolog of polypyrimidine tract binding protein. Mol Cell Biol 2000; 20 : 7463–79. [Google Scholar]
- Kamma H, Portman DS, Dreyfuss G. Cell type-specific expression of hnRNP proteins. Exp Cell Res 1995; 221 : 187–96. [Google Scholar]
- Venables JP, Koh CS, Froehlich U, et al. Multiple and specific mRNA processing targets for the major human hnRNP proteins. Mol Cell Biol 2008; 28 : 6033–43. [Google Scholar]
- Makeyev EV, Zhang J, Carrasco MA, Maniatis T. The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell 2007; 27 : 435–48. [Google Scholar]
- Boutz PL, Chawla G, Stoilov P, Black DL, et al. MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev 2007; 21 : 71–84. [Google Scholar]
- Romano M, Marcucci R, Buratti E, et al. Regulation of 3’ splice site selection in the 844ins68 polymorphism of the cystathionine Beta -synthase gene. J Biol Chem 2002; 277 : 43821–9. [Google Scholar]
- Chou MY, Rooke N, Turck CW, Black DL. hnRNP H is a component of a splicing enhancer complex that activates a c-src alternative exon in neuronal cells. Mol Cell Biol 1999; 19 : 69–77. [Google Scholar]
- Garneau D, Revil T, Fisette JF, Chabot B. Heterogeneous nuclear ribonucleoprotein F/H proteins modulate the alternative splicing of the apoptotic mediator Bcl-x. J Biol Chem 2005; 280 : 22641–50. [Google Scholar]
- Zhou HL, Baraniak AP, Lou H. Role for Fox-1/Fox-2 in mediating the neuronal pathway of calcitonin/calcitonin gene-related peptide alternative RNA processing. Mol Cell Biol 2007; 27 : 830–41. [Google Scholar]
- Ponthier JL, Schluepen C, Chen W, et al. Fox-2 splicing factor binds to a conserved intron motif to promote inclusion of protein 4.1R alternative exon 16. J Biol Chem 2006; 281 : 12468–74. [Google Scholar]
- Zhang W, Liu H, Han K, Grabowski PJ. Region-specific alternative splicing in the nervous system : implications for regulation by the RNA-binding protein NAPOR. RNA 2002; 8 : 671–85. [Google Scholar]
- de la Mata M, Kornblihtt AR. RNA polymerase II C-terminal domain mediates regulation of alternative splicing by SRp20. Nat Struct Mol Biol 2006; 13 : 973–80. [Google Scholar]
- Millhouse S, Manley JL. The C-terminal domain of RNA polymerase II functions as a phosphorylation-dependent splicing activator in a heterologous protein. Mol Cell Biol 2005; 25 : 533–44. [Google Scholar]
- Egloff S, Murphy S. Cracking the RNA polymerase II CTD code. Trends Genet 2008; 24 : 280–8. [Google Scholar]
- De la Mata M, Alonso CR, Kadener S, et al. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 2003; 12 : 525–32. [Google Scholar]
- Roberts GC, Gooding C, Mak HY, et al. Co-transcriptional commitment to alternative splice site selection. Nucleic Acids Res 1998; 26 : 5568–72. [Google Scholar]
- Batsche E, Yaniv M, Muchardt C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat Struct Mol Biol 2006; 13 : 22–9. [Google Scholar]
- Kundu S, Horn PJ, Peterson CL. SWI/SNF is required for transcriptional memory at the yeast GAL gene cluster. Genes Dev 2007; 21 : 997–1004. [Google Scholar]
- Blaustein M, Pelisch F, Srebrow A. Signals, pathways and splicing regulation. Int J Biochem Cell Biol 2007; 39 : 2031–48. [Google Scholar]
- Matter N, Herrlich P, Konig H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 2002; 420 : 691–5. [Google Scholar]
- Boise LH, González-García M, Postema CE, et al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993; 74 : 597–608. [Google Scholar]
- Cloutier P, Toutant J, Shkreta L, et al. Antagonistic effects of the SRp30c protein and cryptic 5’ splice sites on the alternative splicing of the apoptotic regulator Bcl-x. J Biol Chem 2008; 283 : 21315–24. [Google Scholar]
- Paronetto MP, Achsel T, Massiello A, et al. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J Cell Biol 2007; 176 : 929–39. [Google Scholar]
- Massiello A, Roesser JR, Chalfant CE. SAP155 Binds to ceramide-responsive RNA cis-element 1 and regulates the alternative 5’ splice site selection of Bcl-x pre-mRNA. Faseb J 2006; 20 : 1680–2. [Google Scholar]
- Li CY, Chu JY, Yu JK, et al. Regulation of alternative splicing of Bcl-x by IL-6, GM-CSF and TPA. Cell Res 2004; 14 : 473–9. [Google Scholar]
- Revil T, Toutant J, Shkreta L, et al. Protein kinase C-dependent control of Bcl-x alternative splicing. Mol Cell Biol 2007; 27 : 8431–41. [Google Scholar]
- Klinck R, Bramard A, Inkel L, et al. Multiple alternative splicing markers for ovarian cancer. Cancer Res 2008; 68 : 657–63. [Google Scholar]
Liste des figures
|
Figure 1. Les modes d’épissage alternatif. Les modes d’épissage alternatif des pré-ARNm incluent l’exclusion d’exons, l’inclusion mutuellement exclusive d’exons, la concurrence entre sites d’épissage 5’, la concurrence entre sites d’épissage 3’ et la rétention d’introns. |
| Dans le texte | |
|
Figure 2. La reconnaissance des signaux d’épissage. A. La reconnaissance du site d’épissage 5’ (5’ss) (A/CAGGUA/GAGU) s’effectue via le snRNP U1, alors que la reconnaissance du site de branchement de la région riche en pyrimidines et du dinucléotide AG met en jeu respectivement le snRNP U2, U2AF65 et U2AF35. La reconnaissance du site d’épissage 3’ (3’ss) peut être couplée à la liaison du U1 snRNP en aval, contribuant ainsi à la définition de l’exon. B. Des séquences exoniques liées par les protéines SR permettent d’augmenter la reconnaissance des signaux d’épissage par le splicéosome. C. L’interaction entre protéines hnRNP liées près de sites d’épissage distants permet un rapprochement des extrémités de l’intron et contribue à inhiber l’utilisation de pseudo sites introniques. D. La liaison de SRp30c et ASF/SF2 à des régions introniques peut inhiber l’utilisation de sites d’épissage 3’. E. La liaison du régulateur PTB à une séquence intronique contribue à inactiver l’assemblage du spliceosome en inhibant l’interaction entre les snRNP U1 et U2. Lorsqu’il est lié dans l’exon, PTB peut inactiver la définition de l’exon en inhibant le couplage entre U2AF et le snRNP U1. La liaison de la protéine hnRNP A1 à une région exonique peut nuire à l’utilisation du site d’épissage 3’ situé en amont. F. L’expression d’un régulateur pourrait avoir des effets opposés dans différents types cellulaires. Dans l’exemple montré, un effet positif pourrait être attribué au facteur X dans l’épissage d’un exon alternatif pour le type cellulaire A. Cependant, dans le type cellulaire B, l’expression de la protéine X pourrait interférer avec la liaison d’un facteur plus puissant (Y). Le déplacement de Y par X dans le type cellulaire B diminuerait ainsi l’inclusion de l’exon alternatif. |
| Dans le texte | |
|
Figure 3. Couplage entre la transcription et l’épissage. A. Un gène sous le contrôle d’un promoteur (rectangle orange, P1) produit une grande quantité d’ARN pré-messager dans un premier cas (gauche) et, dans d’autres conditions, ce gène exprime une moins grande quantité de transcrits (droite). Une protéine régulatrice (cercle rouge) ayant une activité négative sur un exon alternatif (rectangle vert) de cet ARN pré-messager sera en quantité insuffisante pour contrôler tous les exons alternatifs dans le cas où le transcrit est produit en trop grande quantité. Les rectangles bleus sont des exons constitutifs.B. Tout dépendant lequel des promoteurs est utilisé (rectangle orange ou rectangle mauve), le pré-ARN messager produit pourrait être transporté dans des régions différentes du noyau. Par exemple, lorsque le gène est exprimé sous le contrôle du promoteur 1 (P1, rectangle orange) (partie supérieure), le transcrit est envoyé dans une région riche en protéines régulatrices (cercles verts) et l’exon alternatif (rectangle vert) sera modulé par ces facteurs. Les rectangles bleus sont des exons constitutifs. |
| Dans le texte | |
|
Figure 4. Protéines régulatrices participant à l’épissage alternatif de Bcl-x. L’épissage alternatif du gène Bcl-x engendre une isoforme pro-apoptotique ou anti-apoptotique. La voie de signalisation de la protéine kinase C (PKC) est en cause dans l’inhibition du site d’épissage par l’intermédiaire d’un facteur encore inconnu. La protéine SAP155 inhibe l’utilisation du site d’épissage xS, alors que Sam68, hnRNP A1, F et H favorisent la production de l’isoforme Bcl-xS. SRp30c stimule l’utilisation du site d’épissage 5’ de Bcl-xL. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.