")
")
| Issue |
Med Sci (Paris)
Volume 25, Number 1, Janvier 2009
|
|
|---|---|---|
| Page(s) | 45 - 50 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/200925145 | |
| Published online | 15 janvier 2009 | |
Les deux visages d’ADAM17 dans l’inflammation
Implications dans l’athérosclérose et l’obésité
The two sides of ADAM17 in inflammation: implications in atherosclerosis and obesity
Inserm U626, Faculté de Médecine, 27, boulevard Jean Moulin, 13385 Marseille Cedex 5, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La métalloprotéase ADAM17 (a disintegrin and metalloprotease17) est une protéine transmembranaire de type I, responsable de la protéolyse de nombreux substrats à la surface cellulaire. Sa structure, sa biologie et ses fonctions seront décrites dans cet article, ainsi que son rôle dans l’inflammation et certaines pathologies comme l’athérosclérose et l’obésité. La grande diversité des substrats clivés par ADAM17 et l’extrême complexité de la régulation de sa fonction, laissent entrevoir un domaine de recherche passionnant dont l’exploration aidera à la compréhension des mécanismes physiologiques et physiopathologiques dans lesquels ADAM17 est impliquée.
Abstract
ADAM17 was initially characterized as the TNF Alpha Converting Enzyme (TACE) and, until now, has been the most studied member of the ADAM family. It is a type I transmembrane metalloproteinase involved in the shedding of the extracellular domain of several transmembrane proteins (at least 40) such as cytokines, growth factors, receptors or adhesion molecules. As a consequence, depending on the transmembrane molecule cleaved, one may expect possible opposite effects of ADAM17 activity on inflammation (e.g. TNF and its receptors). The role of ADAM17 in regulating inflammatory cellular processes is clearly demonstrated in cells deficient in active ADAM17 or expressing substrates mutated for the ADAM17 cleavage site. As ADAM17-deficient mice died at birth, mice overexpressing the mutated uncleavable form of some substrates and recently conditional knock-out of ADAM17 are used to approach in vivo the role of this metalloprotease in regulating inflammation. Arguments are provided that ADAM17 plays a role in atherosclerosis, in adipose tissue metabolism, insulin resistance and diabetes. The multitude of substrates cleaved by ADAM17 makes this enzyme an attractive candidate to study its role in inflammation-driven pathologies.
© 2009 médecine/sciences - Inserm / SRMS
La protéolyse de substrats ancrés à la surface cellulaire (shedding) permet aux cellules de libérer des médiateurs et de contrôler leurs interactions avec la matrice extracellulaire et les ligands, qu’ils soient solubles ou associés aux cellules. Ce processus implique des métalloprotéinases qui clivent la partie extracellulaire de molécules transmembranaires (cytokines, récepteurs, molécules d’adhésion et facteurs de croissance). Les métalloprotéinases se répartissent en plusieurs classes : (1) les métalloprotéinases matricielles (MMP) sécrétées (matrilysines, collagénases, gélatinases, stromélysines) ; (2) les métalloprotéinases associées à la membrane par un domaine transmembranaire ou un ancrage GPI (glycosylphosphatidyl inositol) ou membrane-type MMP ; (3) les ADAM (a disintegrin and metalloprotease) qui sont transmembranaires, et (4) les ADAM-TS (TS pour thrombospondin motif) qui sont des ADAM sécrétées. Nous focaliserons cette revue sur ADAM17, la plus étudiée des ADAM, et son implication dans deux pathologies inflammatoires, l’athérosclérose et l’obésité. L’espace limité ne nous permet pas d’aborder le rôle de ADAM17 dans d’autres maladies comme l’arthrite rhumatoïde et le cancer (pour revues, voir [1–3]).
ADAM17 : présentation générale
Les ADAM (environ 40 ont été décrites) appartiennent à la superfamille des métalloprotéinases à zinc [2, 4]. Quatre inhibiteurs protéiques endogènes distincts, les TIMP (tissue inhibitor of metalloproteinase), régulent l’activité de la plupart des ADAM. Les différents domaines structuraux qui composent les ADAM confèrent à certaines d’entre elles des fonctions distinctes dont le clivage protéolytique, l’adhésion et la signalisation. Les ADAM régulent des fonctions cellulaires telles que la prolifération, la fusion cellulaire, et les interactions cellule-matrice et cellule-cellule, ce qui les implique dans différents processus biologiques comme la reproduction et le développement [5]. Leurs rôles dans l’arthrite rhumatoïde, le cancer, l’asthme, l’athérosclérose, l’obésité et le diabète sont de mieux en mieux documentés.
Parmi les ADAM, c’est probablement à l’étude de ADAM17 qu’ont été consacrés le plus de travaux, depuis le clonage de son gène en 1997 [6, 7]. Cette enzyme a été initialement caractérisée comme responsable du clivage du TNFα (tumour necrosis factor), d’où sa dénomination de TNF alpha converting enzyme (TACE), transformant le TNF transmembranaire (tmTNF) de 26 kDa en une forme soluble de 17 kDa (sTNF). Les souris déficientes en activité ADAM17 par mutation du site actif meurent très prématurément, alors que les souris déficientes en TNF sont viables, suggérant qu’ADAM17 clive d’autres molécules que le seul TNF. En effet, l’identification d’autres substrats clivés par ADAM17, comme les ligands de l’EGFR (epidermal growth factor receptor), souligne son rôle prépondérant dans le développement cardiaque et pulmonaire [3]. Au moins une quarantaine de substrats clivés par ADAM17 sont maintenant répertoriés (Figure 1). Les mécanismes moléculaires et enzymatiques à l’origine de cette diversité de sites de clivage ne sont pas encore élucidés. De plus, certains de ces substrats peuvent être clivés par d’autres ADAM (voir la revue d’Edwards et al. [4]), ce qui complique parfois l’interprétation du rôle d’ADAM17 dans certains processus.
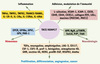 |
Figure 1. Les différents substrats clivés par TACE/ADAM17 et leurs rôles biologiques. La classification proposée n’est pas définitive. En effet, l’implication des substrats d’ADAM17 n’est pas restreinte à une fonction biologique puisque des interactions existent entres les différents processus. Les données sont issues de [4, 42]. APP : amyloid precursor protein ; CSF-1 : colony-stimulating factor 1 ; CSF-1R : colony-stimulating factor 1 receptor ; EPCR : endothelial protein C receptor ; GHR : growth hormone receptor ; GP : glycoprotéine ; HB-EGF : heparin-binding epidermal growth factor ; HER4 : human epidermal growth factor receptor type 4 ; ICAM-1 : intercellular cell adhesion molecule 1 ; IL-15Rα : IL-15 receptor alpha chain ; IL-1Ra : IL-1 receptor antagonist ; IL-1-RII : IL-1 receptor type II ; IL-6Rα : IL-6 receptor alpha chain ; LAG-3 : lymphocyte-activated gene-3 ; NgR : récepteur Nogo ; p75NTR : p75 neurotrophin receptor ; PAR1 : protease activated receptor 1 ; Pref-1 : preadipocyte factor ; PrP : protéine prion ; SorLA : neuronal sorting protein-related receptor ; TGFα : transforming growth factor alpha ; TNFα : tumor necrosis factor alpha ; TNF-R1 : TNF receptor type I (p55) ; TNF-R2 : TNF receptor type II (p75) ; VCAM-1 : vascular cell adhesion molecule 1 ; VEGFR2 : vascular endothelial growth factor receptor type II ; TRANCE/RANKL : tumor necrosis factor-related activation-induced cytokine/receptor κB ligand. |
ADAM17 : structure, expression et activité
Le gène codant ADAM17 a une taille d’environ 55 κB pour un ADNc de 3,5 kB. Il est constitué de 19 exons et est localisé sur les chromosomes 2 chez l’homme et 12 chez la souris. La fonctionnalité de la région promotrice du gène d’ADAM17 a été peu étudiée. Toutefois, chez l’homme, il existe un élément fonctionnel de réponse à l’hypoxie (HRE, hypoxia responsive element). Le niveau des ARNm d’ADAM17 est augmenté dans des tissus pathologiques inflammatoires comme dans l’arthrite rhumatoïde, la cardiomyopathie dilatée, la myocardite ou la colite ulcérative.
ADAM17 est une glycoprotéine transmembranaire de type I synthétisée sous une forme immature, la proforme de ≈100kDa. L’expression d’ADAM17 est ubiquitaire. Chacun des domaines structuraux d’ADAM17 (Figure 2) est directement ou indirectement un régulateur potentiel de l’activité enzymatique. Les interactions que la partie cytoplasmique d’ADAM17 peut établir avec des protéines cytoplasmiques comme SAP97 (synapse-associated protein 97) [8], FHL2 (four and a half lim domain 2) [8, 9] et la kinase ERK [10] contribuent à réguler son activité, son transport et son adressage. La partie cytoplasmique est complétée par un domaine transmembranaire et un domaine extracellulaire. Ce dernier débute par un domaine apparenté à l’EGF suivi d’un domaine riche en cystéines impliqué dans la reconnaissance de certains substrats et l’adhésion tout comme le domaine disintégrine qui suit, liant l’intégrine α5β1. Pour certains auteurs, ce domaine disintégrine faciliterait le rapprochement du substrat avec le site catalytique. Vient ensuite le domaine catalytique, puis le prodomaine terminé par un peptide signal. Le prodomaine agirait à la manière d’une protéine chaperonne permettant à l’enzyme de rester sous sa forme latente inactive protégée de la dégradation protéolytique lors du transport intracellulaire [11]. La séquence RVKR placée entre le domaine catalytique et le prodomaine correspond au site de clivage consensus de la proprotéine convertase furine transformant ADAM17 en une enzyme mature potentiellement active [12]. Le domaine catalytique contient l’ion Zn2+ indispensable à l’activité de l’enzyme. Des changements allostériques lents à proximité du site actif d’ADAM17 contrôleraient l’activité de l’enzyme [13]. TIMP-3 est le seul inhibiteur endogène connu d’ADAM17 [14].
 |
Figure 2. Représentation schématique de la structure d’ADAM17 humaine et de ses fonctions associées. Le domaine cytoplasmique (cytoplasm.) établit des interactions avec des protéines cytoplasmiques modulant le transport, l’adressage et l’activité de l’enzyme. Les domaines apparenté à l’EGF, riche en cystéine (cyst.) et disintégrine, participent à des fonctions d’adhésion et de reconnaissance des substrats. Le domaine métalloprotéase (catalytique) permet à ADAM17 d’acquérir son activité potentielle de clivage, uniquement lorsque la furine le sépare (double flèche) du prodomaine (prodom.). Ce dernier exerce sur l’enzyme une action inhibitrice la maintenant sous forme inactive la protégeant de la dégradation lors de son transport intracellulaire. Parmi les quatre TIMP endogènes, seul TIMP-3 inhibe ADAM17. Les chiffres indiquent la position des aa. |
Bien que des clivages en trans aient été décrits, ADAM17 clive généralement ses substrats en cis. Cela suppose un rapprochement latéral de l’enzyme et de son substrat qui pourrait être favorisé par des changements de fluidité membranaire [15]. Nous avons montré que les radeaux lipidiques membranaires contiennent la forme active d’ADAM17 formant ainsi des zones spécifiques de rencontre entre ADAM17 et certains de ses substrats, et participent aussi à la régulation de leur clivage [16, 43] (Figure 3).
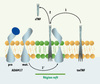 |
Figure 3. Représentation schématique hypothétique de la régulation du clivage du TNF par ADAM17 par compartimentation dans les radeaux lipidiques. La proforme d’ADAM17 est intégrée dans la membrane à l’extérieur des radeaux lipidiques (rafts). Lors de son transport golgien elle est activée par la furine en forme mature (mat.) qui peut être incorporée dans les rafts (1’) (zone verte). La co-localisation d’ADAM17 mature et du tmTNF (1) dans les rafts permet le clivage de ce dernier libérant le sTNF dans le milieu extracellulaire (2). Les rafts représenteraient une plateforme physique de régulation du clivage du TNF par ADAM17. |
ADAM17, inflammation et pathologies à risques cardio-vasculaires
ADAM17 et inflammation
La diversité des substrats clivés par ADAM17 explique que l’enzyme soit un acteur majeur du contrôle de l’inflammation (Figure 1). La dualité d’action d’ADAM17 dans le contrôle de la réaction inflammatoire est illustrée dans le système TNF où le clivage du TNF augmente la disponibilité biologique de la cytokine, alors que le clivage de ses récepteurs entraîne une désensibilisation à l’action de la cytokine.
À côté de l’abondante littérature rapportant les effets du sTNF sur le statut inflammatoire cellulaire, des travaux se sont intéressés à l’effet d’autres substrats clivés par ADAM17 sur le contrôle de la réaction inflammatoire. Nous citerons la forme soluble du récepteur de type II de l’IL(interleukine)-1, considérée comme un antagoniste puissant du système IL-1. De même, la chaîne α du récepteur soluble de l’IL-6 (IL-6Rα), libérée par ADAM17, permet à de nombreux types cellulaires n’exprimant pas l’IL-6Rα, mais possédant la chaîne transductrice du récepteur de l’IL-6 GP130, de répondre à l’IL-6 par un mécanisme de trans-signalisation. ADAM17 pourrait moduler l’adhésion leucocytaire en clivant les molécules d’adhésion à la surface cellulaire comme la chimiokine fractalkine (ou CX3CL1), la L-sélectine, et VCAM-1 (vascular cell adhesion molecule-1).
D’autres approches utilisant des modèles cellulaires exprimant une forme mutée inactive d’ADAM17, des ARN interférents, ou encore des substrats d’ADAM17 mutés non clivables (TNF, Pref-1 [preadipocyte factor 1], HB-EGF [heparin-binding epidermal growth factor], TGFα [transforming growth factor α]) ont permis d’explorer le rôle d’ADAM17. Par exemple, les études consacrées au tmTNF ont permis de documenter ses propriétés de cytotoxicité et d’immunomodulation lorsqu’il est présent à la surface des macrophages et des lymphocytes activés. Le tmTNF exprimé à la surface d’une cellule et liant de manière juxtacrine le récepteur TNFR2 présent sur une autre cellule, exerce une double signalisation conduisant à la production de cytokines immuno-modulatrices : celle qui est déclenchée par le TNFR2 et celle qui est déclenchée dans la cellule elle-même portant le tmTNF selon un mécanisme appelé reverse signaling [17].
Les souris homozygotes déficientes en activité ADAM17 meurent très précocement ce qui a limité les études in vivo de l’implication de l’activité d’ADAM17 sur la composante immuno-inflammatoire et des pathologies associées. Un knock-out conditionnel d’ADAM17 spécifiquement dirigé dans les cellules myéloïdes n’altère cependant pas le développement de la souris, la protège du choc endotoxémique et a permis de montrer le rôle prépondérant de ces cellules dans la libération in vivo du sTNF [18]. Ce n’est que récemment qu’une population de souris déficientes homozygotes en ADAM17 a pu être sélectionnée et maintenue en vie jusqu’à l’âge adulte [19]. Les altérations des populations lymphocytaires présentes chez ces souris (défaut de développement des lymphocytes T et de maturation des lymphcytes B) s’apparentent à celles qui sont observées chez des souris déficientes en TNF. ADAM17 raccourcit la demi-vie de ses substrats à la surface cellulaire ce qui est un obstacle à l’étude in vivo de la fonction de leur forme transmembranaire et donc indirectement de celle d’ADAM17. Pour contourner cet obstacle, des modèles animaux exprimant des formes mutées non clivables de ces substrats ont été construits, et ont permis d’analyser certains aspects de la physiopathologie associée à ADAM17. Ainsi, l’expression ubiquitaire d’un tmTNF non clivable chez la souris se traduit par des modifications de l’immunité innée et acquise responsables de perturbations fonctionnelles d’ordre neurologique, hépatique, immunitaire et métabolique [20–23].
Les souris déficientes en TIMP-3, l’inhibiteur endogène d’ADAM17, sont viables. Elles présentent néanmoins une forte inflammation hépatique, et, lorsqu’elles sont traitées par du lipopolysaccharide, elles développent une inflammation systémique qu’explique la perte du contrôle de l’inhibition d’ADAM17 par TIMP-3 [24]. De même, chez le rat, l’administration de TIMP-3 réduit les dégâts tissulaires hépatiques induits dans un modèle d’ischémie/reperfusion du foie [25].
ADAM17 en pathologie cardiovasculaire
Le chromosome 12 de la souris, qui porte le gène d’ADAM17, contient un locus de susceptibilité à l’athérosclérose. Deux études de cartographie génétique fine, réalisées à partir du croisement de souches susceptibles et résistantes à l’athérosclérose, donnent des résultats divergents sur l’implication d’ADAM17 dans cette pathologie. L’une exclut ADAM17 de ce locus de susceptibilité à l’athérosclérose [26], au contraire de l’autre qui corrèle la résistance à l’athérosclérose à une plus forte expression (mutation sur le promoteur) et une plus forte activité d’ADAM17 [27]. Ce résultat pourrait s’expliquer par la capacité d’ADAM17 à cliver des molécules pro-inflammatoires comme VCAM-1 et la fractalkine. Dans ces deux études, les deux souches croisées ne sont pas identiques et, dans la seconde, le lien entre la résistance à l’athérosclérose et ADAM17 est plus marqué chez la femelle que chez le mâle. Des facteurs épigénétiques et liés au sexe pourraient rendre compte de ces différences. Nos propres résultats montrent, chez la souris mâle génétiquement prédisposée à l’athérosclérose (apoE−/−) et chez l’homme, qu’ADAM17 est exprimée dans les lésions d’athérosclérose (Figure 4) où les macrophages et les microparticules portent cette activité [28, 29]. Ces données sont à rapprocher de celles que nous avons obtenues chez des souris mâles apoE−/− exprimant une forme mutée non clivable du tmTNF. Ces animaux présentent des lésions avancées d’athérosclérose de taille inférieure à celle des souris exprimant le TNF sauvage, alors que la taille des lésions précoces n’est pas significativement différente [30]. Cela suggère un rôle pro-inflammatoire athérogène d’ADAM17 dans les lésions avancées. Ainsi, il est fort probable que l’activité pro- ou anti-athérogène d’ADAM17 résulte du bilan net des substrats pro- et anti-inflammatoires clivés à une période donnée du développement de la pathologie.
 |
Figure 4. Expression d’ADAM17 dans les lésions d’athérosclérose chez la souris. Microphotographie de lésions d’athérosclérose du sinus aortique de souris apoE−/− après 15 semaines de régime gras [28, 29]. A. Coloration des lipides à l’huile rouge O. B. Immunomarquage d’ADAM17 (en rouge) de la zone rectangulaire sélectionnée en A. |
Chez l’homme, bien que le TNF soit un acteur reconnu de la pathologie coronaire et le taux de sTNFR circulant un marqueur prédictif du risque coronaire, peu de données cliniques ont mis en évidence un rôle direct d’ADAM17. ADAM17 serait responsable de l’excès de production de TNF constaté dans les cardiomyopathies dilatées [31]. Dans les monocytes de patients ayant eu un infarctus du myocarde, l’expression et la libération de TNF dépendant d’ADAM17 sont associées aux risques de récidive [32]. Nous avons criblé le gène humain d’ADAM17 et révélé la présence de 19 polymorphismes. Une étude réalisée dans une cohorte de plus de 1 500 patients coronariens (AtheroGene) montre une association positive entre certains de ces polymorphismes, les niveaux de TNF circulant et le syndrome coronaire aigu [33].
ADAM17 dans l’obésité et la résistance à l’insuline
Les territoires adipeux, plus particulièrement la graisse viscérale, sont le siège de réactions inflammatoires qui participent aux complications vasculaires liées à l’obésité et à l’état de résistance à l’insuline [34]. Deux molécules clivées par ADAM17, le TNF et Pref-1 (preadipocyte factor 1), jouent un rôle clé dans le contrôle de l’adipogenèse et de la résistance à l’insuline et illustrent à nouveau la dualité d’action de l’enzyme. Chez l’obèse, le TNF est fortement exprimé dans le tissu adipeux alors que les niveaux plasmatiques du sTNF ne reflètent pas forcément l’importance de l’état inflammatoire dans ce tissu. L’expression du tmTNF est plus élevée dans le tissu adipeux de sujets et souris obèses, conséquence probable d’un clivage diminué, comme cela est montré dans l’adipocyte mature surexprimant le TNF [35]. Dans un modèle murin où l’expression du tmTNF muté non clivable est ciblée dans le tissu adipeux [36], une réduction de 10 % de l’adiposité et de 20 % de la taille des adipocytes est observée en comparaison des souris sauvages. La résistance locale à l’insuline, mais pas la résistance systémique, est augmentée. Selon les auteurs de ces deux études, l’augmentation du tmTNF dans le tissu adipeux serait un mécanisme compensatoire s’opposant à l’augmentation locale de la masse adipeuse induite par le clivage du tmTNF en sTNF. Pref-1 est fortement exprimé dans le préadipocyte et la forme soluble de 50 kDa libérée par ADAM17 (sPref-1) exerce un effet inhibiteur sur la différenciation adipocytaire. Chez la souris, la surexpression ciblée de sPref-1 dans le tissu adipeux entraîne une réduction de la masse adipeuse, et une intolérance au glucose et une résistance à l’insuline systémiques [37]. Ainsi, la disponibilité du TNF et de Pref-1 dans le tissu adipeux exercerait des effets opposés sur la différenciation adipocytaire et l’adiposité, avec des conséquences métaboliques différentes au niveau systémique. Chez des souris haplo-insuffisantes en ADAM17, soumises à un régime gras, on observe une moindre obésité, et de façon concomitante une amélioration des paramètres de la résistance à l’insuline systémique associée à une diminution de la libération de sTNF, IL6Rα et sPref-1 [38]. Cela pourrait indiquer dans ce modèle le rôle prépondérant du TNF par rapport à celui de Pref-1 dans le contrôle de l’adipogenèse et de la résistance à l’insuline. De même, un inhibiteur synthétique d’ADAM17 administré chez des rats hypertendus et insulinorésistants, nourris avec du fructose, mais non obèses, améliore la sensibilité à l’insuline [39]. Les souris déficientes en ADAM17 décrites ci-dessus [19] présentent un phénotype maigre associé à une forte dépense calorique [40]. Une fonction hypothalamique d’ADAM17 contrôlant la balance énergétique est évoquée dans ce travail. De manière intéressante, un lien entre ADAM17, diabète et inflammation vasculaire a été montré chez des souris haplo-déficientes en récepteur à l’insuline. En effet, certaines de ces souris présentent une déficience constitutive en TIMP-3 et développent un diabète. L’excès de sTNF circulant résultant d’une activité exacerbée d’ADAM17 secondaire à la perte de TIMP-3, rendrait compte de l’hyperinsulinémie et de l’augmentation de l’inflammation vasculaire [41]. Ainsi, ADAM17 ferait le lien entre perturbations métaboliques et complications vasculaires qui y sont associées.
Conclusion
ADAM17 est l’un des régulateurs clés du statut inflammatoire dans les pathologies comme l’athérosclérose et l’obésité. Pour autant, l’extrême diversité des substrats pro- et anti-inflammatoires clivés par ADAM17 explique que son rôle au cours de l’évolution de ces maladies soit difficile à cerner. Cette complexité explique probablement pourquoi les inhibiteurs d’ADAM17 n’ont pas encore prouvé leur efficacité clinique même dans des pathologies où l’effet dominant du TNF est reconnu [42]. Une meilleure compréhension des mécanismes associés à la sélectivité de clivage des substrats d’ADAM17 est indispensable pour envisager une pharmacothérapie adaptée à une pathologie précise et aux substrats préférentiellement concernés par cette même pathologie.
Remerciements
Les auteurs remercient l’Inserm, l’Université de la Méditerranée, la Fondation de France et La Fondation pour la Recherche Médicale pour leurs aides financières, et les collaborations de C. Mueller (Berne, Suisse), C. Boulanger, (Inserm U689, Paris), D. Tregouet (Inserm U525, Paris) et les contributions de F. Kopp, B. Bonardo, et N. Saut, aux travaux personnels exposés.
Références
- Mezyk R, Bzowska M, Bereta J. Structure and functions of tumor necrosis factor-alpha converting enzyme. Acta Biochim Pol 2003; 50 : 625–45. [Google Scholar]
- Huovila APJ, Turner AJ, Pelto-Huikko M, et al. Shedding light on ADAM metalloproteinases. Trends Biochem Sci 2005; 30 : 413–22. [Google Scholar]
- Blobel CP. ADAMS : key components in EGFR signalling and development. Nat Rev Mol Cell Biol 2005; 6 : 32–43. [Google Scholar]
- Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med 2008; 29 : 258–89. [Google Scholar]
- Alfandari D, Cousin H, Gaultier A, Darribère T. Les protéines de la famille ADAM : protéolyse, adhérence et signalisation. Med Sci (Paris) 1999; 15 : 1148–51. [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, et al. A metalloproteinase disintegrin that releases tumour necrosis factor-alpha from cells. Nature 1997; 385 : 729–33. [Google Scholar]
- Moss ML, Jin SL, Milla ME, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 1997; 385 : 733–6. [Google Scholar]
- Peiretti F, Deprez-Beauclair P, Bonardo B, et al. Identification of SAP97 as an intracellular binding partner of TACE. J Cell Sci 2003; 116 : 1949–57. [Google Scholar]
- Canault M, Tellier E, Bonardo B, et al. FHL2 interacts with both ADAM-17 and the cytoskeleton and regulates ADAM-17 localization and activity. J Cell Physiol 2006; 208 : 363–72. [Google Scholar]
- Soond SM, Everson B, Riches DWH, Murphy G. ERK-mediated phosphorylation of Thr735 in TNF(alpha)-converting enzyme and its potential role in TACE protein trafficking. J Cell Sci 2005; 118 : 2371–80. [Google Scholar]
- Milla ME, Gonzales PE, Leonard JD. The TACE zymogen : re-examining the role of the cysteine switch. Cell Biochem Biophys 2006; 44 : 342–8. [Google Scholar]
- Peiretti F, Canault M, Deprez-Beauclair P, et al. Intracellular maturation and transport of tumor necrosis factor alpha converting enzyme. Exp Cell Res 2003; 285 : 278–85. [Google Scholar]
- Sagi I, Milla ME. Application of structural dynamic approaches provide novel insights into the enzymatic mechanism of the tumor necrosis factor-(alpha)-converting enzyme. Anal Biochem 2008; 372 : 1–10. [Google Scholar]
- Amour A, Slocombe PM, Webster A, et al. TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett 1998; 435 : 39–44. [Google Scholar]
- Matthews V, Schuster B, Schutze S, et al. Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE). J Biol Chem 2003; 278 : 38829–39. [Google Scholar]
- Tellier E, Canault M, Rebsomen L, et al. The shedding activity of ADAM17 is sequestered in lipid rafts. Exp Cell Res 2006; 312 : 3969–80. [Google Scholar]
- Eissner G, Kolch W, Scheurich P. Ligands working as receptors : reverse signaling by members of the TNF superfamily enhance the plasticity of the immune system. Cytokine Growth Factor Rev 2004; 15 : 353–66. [Google Scholar]
- Horiuchi K, Kimura T, Miyamoto T, et al. Cutting edge : TNF-(alpha)-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol 2007; 179 : 2686–9. [Google Scholar]
- Li N, Boyd K, Dempsey PJ, Vignali DAA. Non-cell autonomous expression of TNF-(alpha)-converting enzyme ADAM17 is required for normal lymphocyte development. J Immunol 2007; 178 : 4214–21. [Google Scholar]
- Akassoglou K, Probert L, Kontogeorgos G, Kollias G. Astrocyte-specific but not neuron-specific transmembrane TNF triggers inflammation and degeneration in the central nervous system of transgenic mice. J Immunol 1997; 158 : 438–45. [Google Scholar]
- Mueller C, Corazza N, Trachsel-Loseth S, et al. Noncleavable transmembrane mouse tumor necrosis factor-alpha (TNF alpha) mediates effects distinct from those of wild-type TNF alpha in vitro and in vivo. J Biol Chem 1999; 274 : 38112–8. [Google Scholar]
- Olleros ML, Guler R, Corazza N, et al. Transmembrane TNF induces an efficient cell-mediated immunity and resistance to Mycobacterium bovis bacillus Calmette-Guerin infection in the absence of secreted TNF and lymphotoxin-(alpha). J Immunol 2002; 168 : 3394–401. [Google Scholar]
- Voros G, Maquoi E, Collen D, Lijnen HR. Differential expression of plasminogen activator inhibitor-1, tumor necrosis factor-(alpha), TNF-(alpha) converting enzyme and ADAMTS family members in murine fat territories. Biochim Biophys Acta 2003; 1625 : 36–42. [Google Scholar]
- Smookler DS, Mohammed FF, Kassiri Z, et al. Cutting edge : tissue inhibitor of metalloproteinase 3 regulates TNF-dependent systemic inflammation. J Immunol 2006; 176 : 721–5. [Google Scholar]
- Tang ZY, Loss G, Carmody I, Cohen AJ. TIMP-3 ameliorates hepatic ischemia/reperfusion injury through inhibition of tumor necrosis factor-alpha-converting enzyme activity in rats. Transplantation 2006; 82 : 1518–23. [Google Scholar]
- Purcell MK, Mu JL, Higgins DC, et al. Fine mapping of Ath6, a quantitative trait locus for atherosclerosis in mice. Mamm Genome 2001; 12 : 495–500. [Google Scholar]
- Holdt LM, Thiery J, Breslow JL, Teupser D. Increased ADAM17 mRNA expression and activity is associated with atherosclerosis resistance in LDL-receptor deficient mice. Arterioscler Thromb Vasc Biol 2008; 28 : 1097–103. [Google Scholar]
- Canault M, Peiretti F, Kopp F, et al. The TNF alpha converting enzyme (TACE/ADAM17) is expressed in the atherosclerotic lesions of apolipoprotein E-deficient mice : possible contribution to elevated plasma levels of soluble TNF alpha receptors. Atherosclerosis 2006; 187 : 82–91. [Google Scholar]
- Canault M, Leroyer AS, Peiretti F, et al. Microparticles of human atherosclerotic plaques enhance the shedding of the tumor necrosis factor-(alpha) converting enzyme/ADAM17 substrates, tumor necrosis factor and tumor necrosis factor receptor-1. Am J Pathol 2007; 171 : 1713–23. [Google Scholar]
- Canault M, Peiretti F, Poggi M, et al. Progression of atherosclerosis in ApoE-deficient mice that express distinct molecular forms of TNF-alpha. J Pathol 2008; 214 : 574–83. [Google Scholar]
- Fedak PWM, Moravec CS, McCarthy PM, et al. Altered expression of disintegrin metalloproteinases and their inhibitor in human dilated cardiomyopathy. Circulation 2006; 113 : 238–45. [Google Scholar]
- Shimoda Y, Satoh M, Nakamura M, et al. Activated tumour necrosis factor-alpha shedding process is associated with in-hospital complication in patients with acute myocardial infarction. Clin Sci (Lond) 2005; 108 : 339–47. [Google Scholar]
- Morange P, Tregouet D, Godefroy T, et al. Polymorphisms of the tumor necrosis factor-alpha (TNF) and the TNF-alpha converting enzyme (TACE/ADAM17) genes in relation to cardiovascular mortality : the Athero Gene study. J Mol Med 2008; 86 : 1153–61. [Google Scholar]
- Juhan-Vague I, Alessi MC, Morange PE. Hypofibrinolysis and increased PAI-1 are linked to atherothrombosis via insulin resistance and obesity. Ann Med 2000; 32 (suppl 1) : 78–84. [Google Scholar]
- Xu H, Uysal KT, Becherer JD, et al. Altered tumor necrosis factor-(alpha) (TNF-[alpha]) processing in adipocytes and increased expression of transmembrane TNF-(alpha) in obesity. Diabetes 2002; 51 : 1876–83. [Google Scholar]
- Xu H, Hirosumi J, Uysal KT, et al. Exclusive action of transmembrane TNF(alpha) in adipose tissue leads to reduced adipose mass and local but not systemic insulin resistance. Endocrinology 2002; 143 : 1502–11. [Google Scholar]
- Lee K, Villena JA, Moon YS, et al. Inhibition of adipogenesis and development of glucose intolerance by soluble preadipocyte factor-1 (Pref-1). J Clin Invest 2003; 111 : 453–61. [Google Scholar]
- Serino M, Menghini R, Fiorentino L, et al. TNF-alpha converting enzyme heterozygous mice are protected from obesity-induced insulin resistance and diabetes. Diabetes 2007; 56 : 2541–6. [Google Scholar]
- Togashi N, Ura N, Higashiura K, et al. Effect of TNF-(alpha)-converting enzyme inhibitor on insulin resistance in fructose-fed rats. Hypertension 2002; 39 : 578–80. [Google Scholar]
- Gelling RW, Yan W, Al-Noori S, et al. Deficiency of TNF(alpha) converting enzyme (TACE/ADAM17) causes a lean, hypermetabolic phenotype in mice. Endocrinology 2008; 149 : 6053–64. [Google Scholar]
- Federici M, Hribal ML, Menghini R, et al. Timp3 deficiency in insulin receptor-haploinsufficient mice promotes diabetes and vascular inflammation via increased TNF-alpha. J Clin Invest 2005; 115 : 3494–505. [Google Scholar]
- Moss ML, Sklair-Tavron L, Nudelman R. Drug insight : tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat Clin Pract Rheumatol 2008; 4 : 300–9. [Google Scholar]
- Surena AL, de Faria GP, Studler JM, et al. DLG1/SAP97 modulates transforming growth factor alpha bioavailability. Biochim Biophys Acta 2008, 27 septembre online. [Google Scholar]
Liste des figures
|
Figure 1. Les différents substrats clivés par TACE/ADAM17 et leurs rôles biologiques. La classification proposée n’est pas définitive. En effet, l’implication des substrats d’ADAM17 n’est pas restreinte à une fonction biologique puisque des interactions existent entres les différents processus. Les données sont issues de [4, 42]. APP : amyloid precursor protein ; CSF-1 : colony-stimulating factor 1 ; CSF-1R : colony-stimulating factor 1 receptor ; EPCR : endothelial protein C receptor ; GHR : growth hormone receptor ; GP : glycoprotéine ; HB-EGF : heparin-binding epidermal growth factor ; HER4 : human epidermal growth factor receptor type 4 ; ICAM-1 : intercellular cell adhesion molecule 1 ; IL-15Rα : IL-15 receptor alpha chain ; IL-1Ra : IL-1 receptor antagonist ; IL-1-RII : IL-1 receptor type II ; IL-6Rα : IL-6 receptor alpha chain ; LAG-3 : lymphocyte-activated gene-3 ; NgR : récepteur Nogo ; p75NTR : p75 neurotrophin receptor ; PAR1 : protease activated receptor 1 ; Pref-1 : preadipocyte factor ; PrP : protéine prion ; SorLA : neuronal sorting protein-related receptor ; TGFα : transforming growth factor alpha ; TNFα : tumor necrosis factor alpha ; TNF-R1 : TNF receptor type I (p55) ; TNF-R2 : TNF receptor type II (p75) ; VCAM-1 : vascular cell adhesion molecule 1 ; VEGFR2 : vascular endothelial growth factor receptor type II ; TRANCE/RANKL : tumor necrosis factor-related activation-induced cytokine/receptor κB ligand. |
| Dans le texte | |
|
Figure 2. Représentation schématique de la structure d’ADAM17 humaine et de ses fonctions associées. Le domaine cytoplasmique (cytoplasm.) établit des interactions avec des protéines cytoplasmiques modulant le transport, l’adressage et l’activité de l’enzyme. Les domaines apparenté à l’EGF, riche en cystéine (cyst.) et disintégrine, participent à des fonctions d’adhésion et de reconnaissance des substrats. Le domaine métalloprotéase (catalytique) permet à ADAM17 d’acquérir son activité potentielle de clivage, uniquement lorsque la furine le sépare (double flèche) du prodomaine (prodom.). Ce dernier exerce sur l’enzyme une action inhibitrice la maintenant sous forme inactive la protégeant de la dégradation lors de son transport intracellulaire. Parmi les quatre TIMP endogènes, seul TIMP-3 inhibe ADAM17. Les chiffres indiquent la position des aa. |
| Dans le texte | |
|
Figure 3. Représentation schématique hypothétique de la régulation du clivage du TNF par ADAM17 par compartimentation dans les radeaux lipidiques. La proforme d’ADAM17 est intégrée dans la membrane à l’extérieur des radeaux lipidiques (rafts). Lors de son transport golgien elle est activée par la furine en forme mature (mat.) qui peut être incorporée dans les rafts (1’) (zone verte). La co-localisation d’ADAM17 mature et du tmTNF (1) dans les rafts permet le clivage de ce dernier libérant le sTNF dans le milieu extracellulaire (2). Les rafts représenteraient une plateforme physique de régulation du clivage du TNF par ADAM17. |
| Dans le texte | |
|
Figure 4. Expression d’ADAM17 dans les lésions d’athérosclérose chez la souris. Microphotographie de lésions d’athérosclérose du sinus aortique de souris apoE−/− après 15 semaines de régime gras [28, 29]. A. Coloration des lipides à l’huile rouge O. B. Immunomarquage d’ADAM17 (en rouge) de la zone rectangulaire sélectionnée en A. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.