")
")
| Issue |
Med Sci (Paris)
Volume 19, Number 12, Décembre 2003
|
|
|---|---|---|
| Page(s) | 1218 - 1225 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/200319121218 | |
| Published online | 15 décembre 2003 | |
C. elegans comme modèle pour les maladies dégénératives héréditaires humaines
C. elegans as a model for human inherited degenerative diseases
1
Laboratoire de Biologie Génomique, Avenir Inserm, Centre d’Études du Polymorphisme Humain, 27, rue Juliette Dodu, 75010 Paris, France
2
Cnrs-CGMC, Université Lyon-1 Claude Bernard, 43, boulevard du 11 Novembre, 69622 Villeurbanne Cedex, France
Résumé
La génétique du nématode C. elegans, modèle majeur en biologie du développement, trouve un champ d’application d’importance croissante pour l’étude de la physiopathologie des maladies humaines. Les modèles obtenus par mutation de gènes conservés chez ce nématode, ou d’animaux transgéniques exprimant des protéines de maladies humaines dont C. elegans est dépourvu, présentent des anomalies retrouvées dans les modèles mammifères. Ces observations suggèrent que les analyses génétiques dans C. elegans permettront d’éclairer la nature des mécanismes cellulaires affectés dans les maladies humaines. Ces modèles sont de plus utiles pour rechercher et caractériser des molécules présentant un potentiel thérapeutique. Cet article illustre ces aspects en prenant pour exemples deux maladies héréditaires, la myopathie de Duchenne et la chorée de Huntington.
Abstract
The nematode C. elegans is an established model for developmental biology. Since the early 90’s, this simple model organism has been increasingly used for studying human disease pathogenesis. C. elegans models based either on the mutagenesis of human disease genes conserved in this nematode or transgenesis with disease genes not conserved in C. elegans show several features that are observed in mammalian models. These observations suggest that the genetic dissection and pharmacological manipulation of disease-like phenotypes in C. elegans will shed light on the cellular mechanisms that are altered in human diseases, and the compounds that may be used as drugs. This review illustrates these aspects by commenting on two inherited degenerative diseases, Duchenne’s muscular dystrophy and Huntington’s neurodegenerative disease.
© 2003 médecine/sciences - Inserm / SRMS
Le nématode Caenorhabditis elegans (C. elegans), modèle majeur pour la biologie du développement [1], permet l’utilisation de l’analyse génétique pour l’étude de l’anatomie et du développement. Grâce au séquençage des génomes et aux techniques qui en sont issues (utilisation de banques de mutants [5], criblages double-hybride dans la levure [6], puces à ADN [7]), C. elegans est entré dans l’ère de la post-génomique [2], comme la levure Saccharomyces cerevisiae [3] ou la mouche Drosophila melanogaster [4]. Mais l’avantage de C. elegans par rapport à ces autres systèmes modèles est que les données sur la fonction des gènes peuvent être interprétées à la lumière de nos connaissances précises sur sa phylogénie cellulaire. Cet organisme invertébré reste en effet le mieux décrit en termes physiologiques, notamment pour l’organisation de son système nerveux.
L’utilisation de C. elegans pour l’étude de la physiopathologie des maladies humaines a commencé vers le milieu des années 1990 par l’étude d’homologues de gènes humains associés à certaines maladies. Un exemple abondamment décrit concerne la susceptibilité génétique à la maladie d’Alzheimer, avec notamment l’étude des présénilines [8] ou l’étude de myopathies comme la myopathie de Duchenne [9]. On peut également étudier les effets de l’introduction de gènes dont C. elegans est dépourvu, comme le gène codant pour la huntingtine, responsable d’une maladie neurodégénérative humaine, la chorée de Huntington. Pour de telles études, C. elegans présente plusieurs avantages : sa transparence, son court cycle de reproduction (3,5 jours) et sa grande facilité de manipulation. À la question de la conservation, chez l’homme, des anomalies et des activités pharmacologiques identifiées chez cet organisme, les réponses sont apportées au cas par cas, avec un pronostic a priori favorable qui provient de la comparaison des gènes - environ 45 % des gènes de C. elegans possèdent au moins un homologue chez l’homme - et des nombreux aspects de sa physiologie cellulaire qui sont bien conservés.
La myopathie de Duchenne
Physiopathologie
La myopathie de Duchenne est une maladie neuromusculaire affectant environ 1 garçon sur 3 500. Elle est due à des mutations dans le gène codant pour la dystrophine, protéine de 420 kDa, identifiée en 1987, localisée sous la membrane de la fibre musculaire, le sarcolemme. Il n’existe pas à ce jour de traitement curatif de la myopathie de Duchenne : la prescription de stéroïdes en continu comme traitement palliatif ralentit l’évolution de la maladie mais comporte de nombreux effets secondaires. Les recherches thérapeutiques sur cette maladie sont rendues difficiles par la lenteur de son évolution, et le fait que, pour des raisons en partie encore inexpliquées, les souris sont peu affectées par des mutations dans ce gène.
De nombreuses études biochimiques effectuées au cours des années 1990 ont montré que la dystrophine est fixée au cytosquelette d’actine par sa partie amino-terminale, et à un complexe protéique transmembranaire nommé le DGC (dystrophin-glycoprotein complex) par sa partie carboxy-terminale. Au fil des années, l’identification des membres de ce complexe n’a que partiellement permis de comprendre la fonction de la dystrophine dans un muscle sain. En revanche, ces études ont montré que plusieurs maladies musculaires dont les symptômes sont proches de ceux de la myopathie de Duchenne sont dues à des mutations dans les gènes de protéines du DGC. L’altération du complexe DGC serait l’événement initial qui provoque la lente dégénérescence musculaire observée chez les patients.
Par quel mécanisme le muscle dégénère-t-il en l’absence de dystrophine ? Cette question reste encore sans réponse et constitue un frein à l’élaboration de traitements de la myopathie de Duchenne et des maladies apparentées. Les hypothèses qui ont été avancées depuis 20 ans sont trop nombreuses pour être rappelées ici, mais on peut les subdiviser en trois classes : (1) la dystrophine contribuerait à la résistance mécanique de la membrane musculaire et son absence serait à l’origine des déchirures de la membrane ; (2) l’absence de dystrophine provoquerait une localisation inappropriée de nombreux composants de la membrane et donc un dysfonctionnement des canaux ioniques, qui, à terme, serait toxique pour la fibre musculaire ; (3) le défaut initial serait plutôt apparenté à un problème de transduction du signal, sans que la nature de ce signal soit actuellement connue. En faveur de cette dernière hypothèse, on observe que différents membres du DGC sont effectivement associés à des protéines impliquées dans la transduction du signal.
Analyses génétiques chez C. elegans
La dystrophine chez C. elegans
Il existe chez C. elegans une protéine apparentée à la dystrophine, dys-1 [9]. Cette protéine est exprimée dans l’ensemble des muscles, et notamment dans les muscles longitudinaux qui sont des muscles striés dont la structure sarcomérique est proche de celle des muscles squelettiques des mammifères. La mutation de ce gène provoque un phénotype d’hyperactivité et d’hypercontraction musculaire dû à une hyperexcitabilité des muscles [9]. En revanche, les mutants dys-1 ne présentent que très peu de dégénérescence musculaire (< 1 % des cellules), peut-être du fait de la courte durée de vie des animaux, le phénotype apparaissant très tardivement chez les mammifères. Mais il est également possible que les muscles de cet animal soient moins sensibles aux perturbations causées par l’absence de dystrophine que ceux des mammifères. Bien que les nématodes et les mammifères aient divergé il y a plusieurs centaines de millions d’années, les fonctions de ce gène ont été en partie conservées puisque l’introduction du gène humain chez les nématodes mutants pour dys-1 peut partiellement restaurer le phénotype sauvage. Par ailleurs, il existe chez C. elegans, outre le gène homologue de la dystrophine, des gènes homologues de la plupart des membres du DGC. Un complexe similaire pourrait donc exister chez cet animal, bien que les données biochimiques à ce sujet soient encore inexistantes.
Identification de gènes fonctionnellement associés à la dystrophine
L’un des atouts de la génétique telle qu’elle se pratique sur les invertébrés est d’associer, sans aucun a priori, un(des) gène(s) à une fonction biologique donnée. Afin d’identifier des protéines fonctionnellement associées à la dystrophine, des cribles génétiques ont été réalisés, à la recherche de mutants présentant le même phénotype que les mutants dys-1. Cette approche repose sur la notion largement admise en génétique que, lorsque des mutations dans des gènes différents provoquent un phénotype similaire, les produits de ces gènes interagissent fonctionnellement. Des animaux ont subi une mutagenèse par l’EMS (éthyl méthanesulfonate), un mutagène chimique qui produit principalement des mutations ponctuelles, et plusieurs dizaines de milliers de descendants ont été analysés. Une vingtaine de nouvelles mutations ont ainsi été isolées. Comme l’indiquent les tests de complémentation, ces mutations sont localisées dans cinq gènes, dont dys-1. L’un de ces gènes code pour la dystrobrévine, une protéine dont il a été démontré chez les mammifères qu’elle est associée à la dystrophine [10]. Chez C. elegans, comme chez les mammifères, les protéines dystrophine et dystrobrévine s’associent par un domaine coiled-coil [11] et cette association est indispensable à la fonction de la dystrobrévine. La surexpression de la dystrobrévine peut légèrement réduire le phénotype induit par la mutation de la dystrophine [12]. Ces résultats ouvrent des perspectives intéressantes, la disparition de la dystrobrévine étant un trait commun à plusieurs maladies myodégénératives dont la myopathie de Duchenne.
Un autre gène identifié par ce crible génétique, dyc-1, code pour une protéine dont l’homologue le plus proche est une protéine de rat nommée CAPON, isolée par un criblage en double hybride lors de la recherche de partenaires de la forme neuronale de la synthase du monoxyde d’azote nNOS. CAPON pourrait être un adaptateur impliqué dans la localisation subcellulaire de nNOS mais cette hypothèse est encore très spéculative [13]. On note cependant avec intérêt que la surexpression de dyc-1 peut, elle aussi, diminuer le phénotype dû à l’absence de dystrophine [14].
Suppression génétique et moléculaire de la dégénérescence musculaire
Dans un contexte génétique où une mutation à faible effet phénotypique du gène myogénique MyoD/hlh-1 a été introduite chez C. elegans, l’absence de dystrophine conduit à une dégénérescence musculaire progressive et à la paralysie des animaux adultes [14]. Près de 30 % des cellules musculaires sont endommagées chez le double mutant dys-1 MyoD/hlh-1 (Figures 1 et 2), qui constitue un outil précieux pour l’isolement de suppresseurs génétiques de la perte musculaire et le criblage de substances chimiques utilisables dans une perspective thérapeutique.
 |
Figure 1. Modèle de dégénérescence musculaire chez le nématode C. elegans : perturbation de l’activité locomotrice par la combinaison de mutations dans les gènes dys-1 et MyoD/hlh-1. Les traces laissées sur le milieu de culture par les animaux sauvages (A), ou mutants MyoD/hlh-1 (B) et dys-1 (C) sont régulières, alors que celles des doubles mutants dys-1 MyoD/hlh-1 du même âge (D) sont irrégulières. Ce défaut est partiellement corrigé par la surexpression du gène dyc-1 (E) (reproduit de [14] avec l’autorisation des Éditions Elsevier). |
 |
Figure 2. Modèle de dégénérescence musculaire chez le nématode C. elegans : défaut de la structure musculaire par la combinaison de mutations dans les gènes dys-1 et MyoD/hlh-1. La musculature révélée par un marquage des fibres d’actine (flèches) est normale chez les animaux sauvages (A) et mutants dys-1 (B) ou MyoD/hlh-1 (C), alors qu’elle est endommagée (flèches) chez le double mutant dys-1 MyoD/hlh-1 (D et E). La dégénérescence musculaire est partiellement corrigée (flèches) par la surexpression du gène dyc-1 (F). |
La recherche de gènes suppresseurs peut se faire au hasard, par mutagenèse sur les animaux double mutants et recherche, dans leur descendance, des animaux présentant une amélioration du phénotype. On peut également effectuer une mutagenèse directe dans des gènes dont on pense qu’ils sont impliqués dans le phénotype obtenu. C’est par exemple le cas du gène egl-19, qui code pour un canal calcique dépendant du potentiel de membrane. On sait que l’absence de dystrophine fonctionnelle chez les patients atteints de myopathie de Duchenne conduit à une concentration de calcium intracellulaire anormalement élevée, délétère à long terme pour le muscle [15]. L’impact de cette augmentation de calcium sur l’évolution de la maladie n’a cependant pas été démontré. La combinaison de mutations dans egl 19, qui diminuent ou augmentent les flux calciques, avec celles du gène de la dystrophine a clairement démontré que la dégénérescence musculaire chez C. elegans est corrélée aux flux calciques [16].
L’existence d’un modèle animal qui mime, même grossièrement, certains aspects de la pathologie humaine, permet de tester l’action de molécules d’intérêt thérapeutique. De tels cribles sont en cours chez C. elegans, dont l’un des avantages est son faible coût par rapport aux modèles mammifères de la maladie. Les composés chimiques qui permettront de réduire le phénotype des mutants de C. elegans seront ensuite testés sur le modèle murin.
La chorée de Huntington
Physiopathologie
La chorée de Huntington est une maladie neurodégénérative dominante qui présente une prévalence d’environ 1 sur 10 000 et qui appartient à la classe des « maladies à polyglutamines ». Cette maladie est en effet due à une expansion de triplets CAG répétés dans la région codante du gène de la huntingtine, conduisant à des insertions de séquences polyglutamines de taille variable (jusqu’à plus de 35 glutamines chez les malades) dans la région amino-terminale de la protéine [17]. Les fragments amino-terminaux de la huntingtine suffisent à induire le dysfonctionnement ou la mort neuronale, probablement du fait d’un repliement anormal de la protéine. L’âge d’apparition de la maladie, d’autant plus précoce que la taille des polyglutamines mutées est grande (phénomène d’anticipation), est largement influencé par le gène de la huntingtine. D’autres gènes pourraient cependant influencer l’âge d’apparition de la maladie, en agissant avec une plus faible pénétrance, c’est-à-dire dans des groupes restreints de malades [18]. Du point de vue neuropathologique, la chorée de Huntington se traduit par une dégénérescence sélective des neurones du noyau caudé et du néocortex. Les neurones survivants présentent souvent des neurites dystrophiques contenant des agrégats, ainsi que des inclusions nucléaires enrichies en huntingtine et en ubiquitine. Il n’existe pas de traitement curatif de cette maladie qui peut évoluer sur plusieurs dizaines d’années avec un âge d’apparition moyen autour de 40 ans.
La huntingtine est une protéine ubiquitaire principalement cytoplasmique, associée au cytosquelette et à diverses structures cellulaires (vésicules à clathrine, endosomes, vésicules synaptiques). Elle contient une région riche en proline qui interagit avec plusieurs protéines à domaines spécifiques hydrophobes comme les domaines WW ou SH3. Chez la drosophile, la huntingtine ne contient ni domaines polyglutamines ni régions riches en proline. Des progrès notables dans la compréhension des mécanismes de la maladie de Huntington ont été réalisés grâce au développement de modèles biochimiques, cellulaires, génétiques et lésionnels [21–23]. En particulier, les phénotypes d’inactivation du gène codant pour la huntingtine chez la souris suggèrent que la protéine serait impliquée dans la neurogenèse [19] et dans la survie neuronale à l’âge adulte [20]. Les propriétés toxiques induites par la huntingtine mutée et les « pertes de fonction » résultant de l’absence de la protéine normale sont d’une grande diversité : anomalies du trafic et de la dégradation des protéines, anomalies de la transduction du signal [24–26] et anomalies de la transcription [27] provoquant éventuellement des pertes d’expression de neurotrophines comme le BDNF (brain-derived neurotrophic factor) [28] ou le NGF (nerve growth factor) [29]. Certains aspects de la maladie restent cependant sujet à controverse, comme par exemple l’éventuelle toxicité des agrégats, structures hétérogènes et réversibles [30], par rapport à celle des protéines mutantes solubles [31]. Les agrégats pourraient agir par séquestration de facteurs de transcription à polyglutamines comme CBP (CREB binding protein), susceptibles d’être recrutés dans les inclusions nucléaires par interaction avec les polyglutamines de la huntingtine mutée [27]. Les formes solubles nucléaires ou cytoplasmiques de la huntingtine mutée agiraient pour leur part au travers de liaisons plus fortes ou plus faibles que la normale avec des protéines partenaires, parmi lesquelles des facteurs de transcription [32] et des protéines de la transduction du signal [25, 33, 34].
Modélisation chez C. elegans
Huntingtine et protéome de C. elegans
Le nématode C. elegans étant dépourvu de huntingtine, on peut se demander si l’expression transgénique de cette protéine dans les neurones de C. elegans va produire un phénotype neurologique pertinent pour l’étude de la physiopathologie de la chorée de Huntington. En faveur de cette hypothèse, on observe que 13 des 30 partenaires de la huntingtine sont conservés chez le nématode et trouvés dans toutes les catégories fonctionnelles associées à la chorée de Huntington (Figure 3). D’ailleurs, des tests de liaison in vitro indiquent que la huntingtine est physiologiquement capable d’interagir avec le protéome de C. elegans [18]. En revanche, l’absence de domaines polyglutamines chez les homologues de CBP de C. elegans ne permet pas la reproduction du phénomène de séquestration chez les nématodes transgéniques exprimant la huntingtine mutée.
 |
Figure 3. Conservation des protéines partenaires de la huntingtine chez Drosophila melanogaster, C. elegans, et Saccharomyces cerevisiae. Ce schéma incorpore 30 protéines partenaires de la huntingtine identifiées essentiellement par des tests en double hybride chez la levure, et parfois confirmées par des tests d’interaction in vitro ou de co-localisation in vivo. Parmi ces protéines, 23 sont conservées chez la drosophile (F), 16 chez le nématode (W), et 6 chez la levure (Y). Les 16 protéines conservées chez C. elegans sont réparties dans plusieurs catégories fonctionnelles : facteurs de transcription (fond violet), transduction du signal (fond jaune), métabolisme (fond bleu), système ubiquitine-protéasome (fond orange), cytosquelette/endocytose (fond gris). Les protéines dont le rôle biologique est inconnu sont indiquées par un fond blanc. La présence d’un domaine glutamine-alanine (polyQA), polyglutamine polyQ (cadre vert), WW (cadre rouge), ou SH3 (cadre bleu) est indiquée. Les alignements ont été calculés avec les logiciels BLASTP (E < 10-20) et Smith-Waterman (S > 300). |
Analyses génétiques de la toxicité neuronale
Le premier modèle décrit de toxicité neuronale des polyglutamines chez C. elegans [35] a été obtenu par l’expression d’un fragment amino-terminal de la huntingtine mutée sous le contrôle du promoteur osm-10, spécifique des neurones sensoriels ASH impliqués dans la perception osmotique et dans la réponse au toucher au niveau du nez de l’animal (Figure 4). Une séquence de 150 glutamines produit un dysfonctionnement neuronal et une mort cellulaire à faible pénétrance. Dans ce modèle, le fragment amino-terminal de la huntingtine mutée est détecté sous forme d’agrégats périnucléaires, sans inclusions dans le noyau. Des résultats identiques ont été obtenus lors de l’expression d’un court fragment de la huntingtine mutée, correspondant approximativement à l’exon 1, fusionné à une protéine fluorescente, la CFP (cyan fluorescent protein) et exprimé, sous contrôle du promoteur mec-3, dans plusieurs des neurones mécanosensoriels [33] (Figure 5).
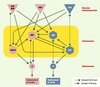 |
Figure 4. Circuits neuronaux testés dans le modèle de dysfonctionnement neuronal induit par l’expression des polyglutamines dans les neurones mécanosensoriels chez C. elegans. Ce schéma montre les six neurones mécanosensoriels (ALM, AVM, deux cellules ASH, et deux cellules PLM au niveau de la queue de l’animal) dans lesquels le promoteur du gène mec-3 est actif. Ces neurones sont connectés aux interneurones qui relaient les signaux vers les motoneurones A et B. Le circuit neuronal plus spécifiquement mis en jeu lors d’une stimulation des neurones PLM localisés au niveau de la queue et qui provoque un déplacement en avant de l’animal est indiqué en bleu. |
Dans cette expérience, la morphologie des neurones est révélée par l’expression de la YFP (yellow fluorescent protein) sous contrôle du promoteur du gène mec-7 codant pour la b-tubuline chez C. elegans (Figure 5, A et B). L’expression des polyglutamines mutées produit un important dysfonctionnement neuronal mais pas de mort cellulaire. La majorité des animaux transgéniques exprimant les polyglutamines mutées ne répondent plus au toucher au niveau de la queue (phénotype Mec postérieur). Ce phénotype est testé manuellement en effleurant l’animal avec un cil monté sur un cure-dent. S’il peut paraître rudimentaire, ce test n’en est pas moins très sensible, étant fondé sur des comportements reproductibles contrôlés par la seule paire de neurones PLM. Le phénotype Mec postérieur est partiellement corrélé à la présence d’agrégats et d’anomalies morphologiques des axones (Figure 5F). L’existence d’un phénotype de dysfonctionnement neuronal associé à l’expression de polyglutamines chez C. elegans devrait permettre l’analyse génétique de ce qui pourrait être une étape précoce de la chorée de Huntington, et la recherche ultérieure de suppresseurs génétiques. À terme, ces suppresseurs pourront être comparés aux suppresseurs - obtenus chez la drosophile - de la mort neuronale induite par les polyglutamines [36, 37].
 |
Figure 5. Modèle de dysfonctionnement neuronal sans mort cellulaire chez C. elegans. Anomalies induites par l’expression de la partie amino-terminale de la huntingtine normale (19 glutamines [gln], C, D) ou mutée (128 glutamines, E, F) fusionnée à la CFP (cyan fuorescent protein) dans les neurones mécanosensoriels de la queue de l’animal. La morphologie des neurones est révélée par l’expression diffuse de la YFP (yellow fluorescent protein) dans le corps cellulaire (A, C, E) et l’axone (B, D, F) sous contrôle du promoteur du gène mec-7 codant pour la b-tubuline chez C. elegans (coloration en vert). C à F : profil d’agrégration de la huntingtine fusionnée à la CFP et exprimée sous contrôle du promoteur du gène mec-3 codant pour un facteur de transcription exprimé dans 10 neurones de C. elegans, dont les cellules PLM (coloration en orange). La formation d’agrégats périnucléaires est observée aussi bien pour la huntingtine normale (C) que mutée (E), et n’est donc pas corrélée au dysfonctionnement spécifiquement observé avec la huntingtine mutée. En revanche, la huntingtine mutée provoque l’apparition d’agrégats (F : flèche bleue) et d’anomalies morphologiques (F : flèches blanches) des axones alors que la huntingtine normale n’induit pas ces effets de façon significative (D). |
Manipulations pharmacologiques
Dans le domaine des maladies neurodégénératives, l’identification de substances chimiques capables de corriger le défaut physiologique résultant de mutations génétiques peut être une alternative aux thérapies substitutives par réintroduction du gène non muté (thérapie génique). Dans un premier temps, les organismes modèles simples comme C. elegans, de manipulation rapide et peu coûteuse, peuvent permettre de sélectionner un nombre limité de molécules qui pourront ensuite être testées chez la souris. Une autre approche consiste à cribler des chimiothèques, puis à caractériser le mode d’action des composés actifs. Des modèles cellulaires [26, 38] et des modèles murins [39, 40] ou de drosophile [41] ont déjà permis d’identifier un certain nombre de composés à potentiel thérapeutique comme les inhibiteurs des histone désacétylases (effet sur la transcription), la minocycline (effet sur les caspases), et la créatine ou l’IGF1 (insulin-like growth factor 1) (effets neuroprotecteurs). Le modèle C. elegans de toxicité des polyglutamines [33] est actuellement utilisé dans le cadre d’un programme international de recherche de molécules thérapeutiques sous l’égide du National Institute of Neurodegenerative Diseases and Stroke aux États-Unis. Ce programme, fondé sur une collection de 1 040 médicaments testés dans plus de 15 modèles « polyglutamines » non mammifères [42–44], a permis de comparer la valeur informative de C. elegans et d’autres organismes modèles simples pour les maladies neurodégénératives. Le modèle C. elegans s’avère très sensible et pourrait servir de guide, avec un petit nombre d’autres modèles, pour la sélection préliminaire des classes de composés à évaluer en priorité dans les phases ultérieures de ce programme.
Conclusions et perspectives
Les modèles C. elegans fondées sur la mutagenèse de gènes de maladies humaines conservés chez ce nématode ont permis de révéler de nouveaux aspects de la physiopathologie de maladies dégénératives humaines comme la myopathie de Duchenne ou la maladie d’Alzheimer. Développés plus récemment, les modèles de maladies humaines fondés sur l’expression de protéines non conservées chez C. elegans - situation fréquente pour les maladies neurodégénératives héréditaires - semblent prometteurs. Chez la drosophile, les modèles transgéniques ont permis de confirmer le rôle de protéines dont on suspectait déjà l’implication [45]. Le fait que plusieurs protéines de maladies neurodégénératives soient pathogènes chez C. elegans ou chez la drosophile suggèrent que leur toxicité n’est pas spécifique des neurones de mammifère. Si les mécanismes de la toxicité neuronale induite par les polyglutamines mutées chez C. elegans sont les mêmes que chez l’homme, nous pourrons savoir s’ils relèvent d’une toxicité intrinsèque des polyglutamines, éventuellement commune à plusieurs maladies, ou s’ils permettront d’étudier d’autres aspects plus complexes des maladies à polyglutamines.
Références
- Felix MA, Labouesse M, Ségalat L. Caenorhabditis elegans, un organisme modèle en biologie. Paris : Éditions Hermann, 2002 : 194 p. [Google Scholar]
- The C. elegans sequencing consortium. Genome sequence of the nematode C. elegans : a platform for investigating biology. Science 1998; 282 : 2012–8. [Google Scholar]
- Goffeau A, Barrell BG, Bussey H, et al. Life with 6 000 genes. Science 1996; 274 : 63–7. [Google Scholar]
- Adams MD, Celniker SE, Holt RA, et al. The genome sequence of Drosophila melanogaster. Science 2000; 287 : 2185–95. [Google Scholar]
- Jansen G. Gene inactivation in Caenorhabditis elegans. Curr Genomics 2002; 3 : 59–68. [Google Scholar]
- Rual JF, Lamesh P, Vandenhaute J, Vidal M. The Caenorhabditis elegans interactome mapping project. Curr Genomics 2002; 3 : 83–94. [Google Scholar]
- Reinke V. Defining development through gene expression profiling. Curr Genomics 2002; 3 : 95–111. [Google Scholar]
- Baumeister R. The physiological role of presenilins in cellular differentiation : lessons from model organisms. Eur Arch Psychiatr Clin Neurosci 1999; 249 : 280–7. [Google Scholar]
- Bessou C, Giugia JB, Franks CJ, Holden-Dye L, Segalat, L. Mutations in the Caenorhabditis elegans dystrophin-like gene dys-1 lead to hyperactivity and suggest a link with cholinergic transmission. Neurogenetics 1998; 2 : 61–72. [Google Scholar]
- Gieseler K, Bessou C, Segalat L. Dystrobrevin- and dystrophin-like mutants display similar phenotypes in the nematode Caenorhabditis elegans. Neurogenetics 1999; 2 : 87–90. [Google Scholar]
- Gieseler K, Mariol MC, Bessou C, et al. Molecular, genetic and physiological characterisation of dystrobrevin-like (dyb-1) mutants of Caenorhabditis elegans. J Mol Biol 2001; 307 : 107–17. [Google Scholar]
- Gieseler K, Grisoni K, Mariol MC, Segalat L. Overexpression of dystrobrevin delays locomotion defects and muscle degeneration in a dystrophin-deficient Caenorhabditis elegans. Neuromusc Disord 2002; 12 : 371–7. [Google Scholar]
- Jaffrey SR, Benfenati F, Snowman AM, Czernik AJ, Snyder SH. Neuronal nitric-oxide synthase localization mediated by a ternary complex with synapsin and CAPON. Proc Natl Acad Sci USA 2002; 99 : 3199–204. [Google Scholar]
- Gieseler K, Grisoni K, Segalat L. Genetic suppression of phenotypes arising from mutations in dystrophin-related genes in Caenorhabditis elegans. Curr Biol 2000; 10 : 1092–7. [Google Scholar]
- Mallouk N, Jacquemond V, Allard B. Elevated subsarcolemmal Ca2+ in mdx mouse skeletal muscle fibers detected with Ca2+-activated K+ channels. Proc Natl Acad Sci USA 2000; 97 : 4950–5. [Google Scholar]
- Mariol MC, Segalat L. Muscular degeneration in the absence of dystrophin is a calcium-dependent process. Curr Biol 2001; 11 : 1691–4. [Google Scholar]
- The Huntington’s disease collaborative research group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993; 72 : 971–83. [Google Scholar]
- Holbert S, Denghien I, Kiechle T, et al. The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin : neuropathologic and genetic evidence for a role in Huntington’s disease pathogenesis. Proc Natl Acad Sci USA 2001; 98 : 1811–6. [Google Scholar]
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet 1995; 11 : 155–63. [Google Scholar]
- Dragatsis I, Levine MS, Zeitlin S. Inactivation of hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet 2000; 26 : 300–6. [Google Scholar]
- Sipione S, Cattaneo E. Modeling Huntington’s disease in cells, flies, and mice. Mol Neurobiol 2001; 23 : 21–51. [Google Scholar]
- Menalled LB, Chesselet MF. Mouse models of Huntington’s disease. Trends Pharmacol Sci 2002; 23 : 32–9. [Google Scholar]
- Rubinsztein DC. Lessons from animal models of Huntington’s disease. Trends Genet 2002; 18 : 202–9. [Google Scholar]
- Liu YF, Deth RC, Devys D. SH3 domain-dependent association of huntingtin with epidermal growth factor receptor signaling complexes. J Biol Chem 1997; 272 : 8121–4. [Google Scholar]
- Song C, Perides G, Liu YF. Expression of full-length polyglutamine-expanded huntingtin disrupts growth factor receptor signaling in rat pheochromocytoma (PC12) cells. J Biol Chem 2002; 277 : 6703–7. [Google Scholar]
- Humbert S, Bryson EA, Cordelières FP, et al. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves huntingtin phosphorylation by Akt. Dev Cell 2002; 2 : 831–7. [Google Scholar]
- Nucifora FC Jr, Sasaki M, Peters MF, et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 2001; 291 : 2423–8. [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001; 293 : 493–8. [Google Scholar]
- Li SH, Cheng AL, Zhou H, et al. Interaction of Huntington disease protein with transcriptional activator sp1. Mol Cell Biol 2002; 22 : 1277–87. [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 2000; 101 : 57–66. [Google Scholar]
- Sisodia SS. Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? Cell 1998; 95 : 1–4. [Google Scholar]
- Yu ZX, Li SH, Nguyen HP, Li XJ. Huntingtin inclusions do not deplete polyglutamine-containing transcription factors in HD mice. Hum Mol Genet 2002; 11 : 905–14. [Google Scholar]
- Parker JA, Connolly JB, Wellington C, et al. Expanded polyglutamines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc Natl Acad Sci USA 2001; 98 : 13318–23. [Google Scholar]
- Li SH, Li H, Torre ER, Li XJ. Expression of huntingtin-associated protein-1 in neuronal cells implicates a role in neuritic growth. Mol Cell Neurosci 2000; 16 : 168–83. [Google Scholar]
- Faber PW, Alter JR, MacDonald ME, Hart AC. Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc Natl Acad Sci USA 1999; 96 : 179–84. [Google Scholar]
- Fernandez-Funez P, Nino-Rosales ML, De Gouyon B, et al. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature 2000; 408 : 101–6. [Google Scholar]
- Higashiyama H, Hirose F, Yamaguchi M, et al. Identification of ter94, Drosophila VCP, as a modulator of polyglutamine-induced neurodegeneration. Cell Death Differ 2002; 9 : 264–73. [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 1998; 95 : 55–66. [Google Scholar]
- Chen M, Ona VO, Li M, et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 2000; 6 : 797–801. [Google Scholar]
- Ferrante RJ, Andreassen OA, Jenkins BG, et al. Neuroprotective effects of creatine in a transgenic mouse model of Huntington’s disease. J Neurosci 2000; 20 : 4389–97. [Google Scholar]
- Steffan JS, Bodai L, Pallos J, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 2001; 413 : 739–43. [Google Scholar]
- Abbott A. Neurologists strike gold in drug screen effort. Nature 2002; 417 : 109. [Google Scholar]
- Heemskerk J, Tobin AJ, Bain LJ. Teaching old drugs new tricks. Meeting of the neurodegeneration drug screening consortium, 7-8 April 2002, Washington, DC, USA. Trends Neurosci 2002; 25 : 494–6. [Google Scholar]
- Heemskerk J, Tobin AJ, Ravina B. From chemical to drug: neurodegeneration drug screening and the ethics of clinical trials. Nat Neurosci 2002; 5 (suppl 1) : 1027–9. [Google Scholar]
- Link CD. Transgenic invertebrate models of age-associated neurodegenerative diseases. Mech Ageing Dev 2001; 122 : 1639–49. [Google Scholar]
Liste des figures
|
Figure 1. Modèle de dégénérescence musculaire chez le nématode C. elegans : perturbation de l’activité locomotrice par la combinaison de mutations dans les gènes dys-1 et MyoD/hlh-1. Les traces laissées sur le milieu de culture par les animaux sauvages (A), ou mutants MyoD/hlh-1 (B) et dys-1 (C) sont régulières, alors que celles des doubles mutants dys-1 MyoD/hlh-1 du même âge (D) sont irrégulières. Ce défaut est partiellement corrigé par la surexpression du gène dyc-1 (E) (reproduit de [14] avec l’autorisation des Éditions Elsevier). |
| Dans le texte | |
|
Figure 2. Modèle de dégénérescence musculaire chez le nématode C. elegans : défaut de la structure musculaire par la combinaison de mutations dans les gènes dys-1 et MyoD/hlh-1. La musculature révélée par un marquage des fibres d’actine (flèches) est normale chez les animaux sauvages (A) et mutants dys-1 (B) ou MyoD/hlh-1 (C), alors qu’elle est endommagée (flèches) chez le double mutant dys-1 MyoD/hlh-1 (D et E). La dégénérescence musculaire est partiellement corrigée (flèches) par la surexpression du gène dyc-1 (F). |
| Dans le texte | |
|
Figure 3. Conservation des protéines partenaires de la huntingtine chez Drosophila melanogaster, C. elegans, et Saccharomyces cerevisiae. Ce schéma incorpore 30 protéines partenaires de la huntingtine identifiées essentiellement par des tests en double hybride chez la levure, et parfois confirmées par des tests d’interaction in vitro ou de co-localisation in vivo. Parmi ces protéines, 23 sont conservées chez la drosophile (F), 16 chez le nématode (W), et 6 chez la levure (Y). Les 16 protéines conservées chez C. elegans sont réparties dans plusieurs catégories fonctionnelles : facteurs de transcription (fond violet), transduction du signal (fond jaune), métabolisme (fond bleu), système ubiquitine-protéasome (fond orange), cytosquelette/endocytose (fond gris). Les protéines dont le rôle biologique est inconnu sont indiquées par un fond blanc. La présence d’un domaine glutamine-alanine (polyQA), polyglutamine polyQ (cadre vert), WW (cadre rouge), ou SH3 (cadre bleu) est indiquée. Les alignements ont été calculés avec les logiciels BLASTP (E < 10-20) et Smith-Waterman (S > 300). |
| Dans le texte | |
|
Figure 4. Circuits neuronaux testés dans le modèle de dysfonctionnement neuronal induit par l’expression des polyglutamines dans les neurones mécanosensoriels chez C. elegans. Ce schéma montre les six neurones mécanosensoriels (ALM, AVM, deux cellules ASH, et deux cellules PLM au niveau de la queue de l’animal) dans lesquels le promoteur du gène mec-3 est actif. Ces neurones sont connectés aux interneurones qui relaient les signaux vers les motoneurones A et B. Le circuit neuronal plus spécifiquement mis en jeu lors d’une stimulation des neurones PLM localisés au niveau de la queue et qui provoque un déplacement en avant de l’animal est indiqué en bleu. |
| Dans le texte | |
|
Figure 5. Modèle de dysfonctionnement neuronal sans mort cellulaire chez C. elegans. Anomalies induites par l’expression de la partie amino-terminale de la huntingtine normale (19 glutamines [gln], C, D) ou mutée (128 glutamines, E, F) fusionnée à la CFP (cyan fuorescent protein) dans les neurones mécanosensoriels de la queue de l’animal. La morphologie des neurones est révélée par l’expression diffuse de la YFP (yellow fluorescent protein) dans le corps cellulaire (A, C, E) et l’axone (B, D, F) sous contrôle du promoteur du gène mec-7 codant pour la b-tubuline chez C. elegans (coloration en vert). C à F : profil d’agrégration de la huntingtine fusionnée à la CFP et exprimée sous contrôle du promoteur du gène mec-3 codant pour un facteur de transcription exprimé dans 10 neurones de C. elegans, dont les cellules PLM (coloration en orange). La formation d’agrégats périnucléaires est observée aussi bien pour la huntingtine normale (C) que mutée (E), et n’est donc pas corrélée au dysfonctionnement spécifiquement observé avec la huntingtine mutée. En revanche, la huntingtine mutée provoque l’apparition d’agrégats (F : flèche bleue) et d’anomalies morphologiques (F : flèches blanches) des axones alors que la huntingtine normale n’induit pas ces effets de façon significative (D). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.