")
")
| Issue |
Med Sci (Paris)
Volume 18, Number 6-7, Juin–Juillet 2002
|
|
|---|---|---|
| Page(s) | 727 - 736 | |
| Section | M/S Revues : Articles de Synthèse | |
| DOI | https://doi.org/10.1051/medsci/20021867727 | |
| Published online | 15 juin 2002 | |
La maladie d’Alzheimer : une tauopathie parmi d’autres ?
Alzheimer’s disease: just another tauopathy?
Inserm U.422, nstitut de Médecine Prédictive et Recherche Thérapeutique, place de Verdun, 59045 Lille Cedex, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La dégénérescence neurofibrillaire - agrégation intraneuronale de protéines tau anormalement phosphorylées - est un processus dégénératif qui affecte la région hippocampique des personnes âgées. Dans la maladie d’Alzheimer, la dégénérescence neurofibrillaire peut progresser vers les régions corticales associatives, selon un schéma établi, séquentiel et hiérarchique. À ce stade, les dépôts amyloïdes sont importants. Pour comprendre la maladie, il faut être capable de différencier la cause de la conséquence. Les formes familiales de la maladie d’Alzheimer ont mis en lumière le rôle majeur joué par le peptide β-amyloïde. Cependant, au cours des dernières années, une pièce maîtresse est venue modifier notre compréhension globale : un grand nombre de maladies neurodégénératives sont dues à des altérations directe ou indirecte du métabolisme de la protéine tau. Nous montrons ici que la maladie d’Alzheimer est une véritable « tauopathie », qui ne peut toutefois se développer qu’en présence d’un dysfonctionnement de l’APP, précurseur du peptide β-amyloïde.
Abstract
Tau proteins belong to the family of microtubule-associated proteins. They are mainly expressed in neurons where they play an important role in the assembly of tubulin monomers into microtubules to constitute the neuronal microtubule network. Tau proteins are translated from a single gene located on chromosome 17. Their expression is developmentally regulated by an alternative splicing mechanism and six different isoforms exist in the human adult brain. Tau proteins are the major constituents of fibrillar lesions described in Alzheimer’s disease and numerous neurodegenerative disorders referred to as « tauopathies ». Molecular analysis has revealed that an abnormal phosphorylation might be one of the important events in the process leading to their aggregation. Moreover, a specific set of pathological tau proteins exhibiting a typical biochemical pattern, and a different regional and laminar distribution could characterize each of these disorders. Finally, the recent discovery of tau gene mutations in fron-totemporal dementia with parkinsonism linked to chromosome 17 has reinforced the direct role attributed to tau proteins in the pathogenesis of neurodegenerative disorders, and underlined the fact that distinct sets of tau isoforms expressed in different neuronal populations could lead to different pathologies. Based on these findings, new animal and cell models are developed and should allow for a better understanding of neurofibrillary degeneration. ◊
© 2002 médecine/sciences - Inserm / SRMS
La maladie d’Alzheimer, décrite au début du siècle (1907) par Aloïs Alzheimer [1], est une maladie neurodégénérative liée à l’âge, caractérisée par un syndrome démentiel et par la présence, dans le cortex cérébral, de lésions neuropathologiques particulières : des dépôts amyloïdes et des neurones subissant ce qu’on dénomme un processus de « dégénérescence neurofibrillaire ». Au milieu des années 1980, les caractéristiques biochimiques de ces deux lésions ont été définies : les dépôts amyloïdes sont constitués d’un peptide de 40 à 42 acides aminés appelé Aβ [2] et les dégénérescences neurofibrillaires intraneuronales résultent de l’agrégation de protéines microtubulaires tau [3]. La biologie moléculaire a permis d’établir une classification de la plupart des maladies neurodégénératives. Ainsi, la maladie d’Alzheimer et de nombreuses autres affections ont été regroupées sous le terme de « tauopathies » car elles présentent toutes des agrégats intracellulaires de protéines tau (→). En revanche, la maladie de Parkinson et les autres maladies dans lesquelles sont retrouvés des agrégats intracellulaires d’α-synucléine (→→) ont été dénommées α-synucléinopathies.
(→) m/s 1997, n°8-9, p. 989
(→→) m/s 2000, n°8-9, p. 956
Dans la maladie d’Alzheimer, la dégénérescence neurofibrillaire (DNF) n’intéresse que certains groupes particuliers de neurones, principalement des neurones pyramidaux. La dégénérescence neurofibrillaire correspond à une accumulation intraneuronale de fibrilles formées de filaments très caractéristiques, appelés paires de filaments appariés en hélice (PHF, paired helical filaments). Ces filaments pathologiques sont d’excellents marqueurs ultrastructuraux du processus dégénératif de type Alzheimer. Les PHF sont également observés dans les neurites en dégénérescence qui abondent dans le neuropile et dans les plaques neuritiques. Les PHF sont constitués par l’assemblage de protéines microtubulaires tau. En immunohistochimie, les neurones en dégénérescence neurofibrillaire sont d’abord reconnus par des anticorps dirigés contre la protéine tau [3]. Un marquage positif peut aussi être observé avec des anticorps dirigés, entre autres, contre l’apolipoprotéine E, des protéines du cycle cellulaire (kinases dépendantes de cyclines-Cdk, polo-kinases, Cdc25, prolyl-peptidyl cis/trans isomérase-Pin1), les glycosaminoglycanes/protéoglycanes, les neurofilaments et l’ubiquitine peuvent également marquer les neurones en dégénérescence neurofibrillaire.
Tauopathies et agrégats de protéines tau
La dégénérescence neurofibrillaire n’est pas une lésion histologique spécifique de la maladie d’Alzheimer. Des agrégats de protéines tau sont retrouvés dans le cerveau de patients atteints de multiples affections : la trisomie 21, de nombreux syndromes parkinsoniens (dégénérescence cortico-basale, paralysie supranucléaire progressive [PSP], maladie de Parkinson post-encéphalitique, syndrome de l’île de Guam), certaines démences fronto-temporales (DFT) telles que la maladie de Pick et les démences fronto-temporales avec syndrome parkinsonien liées au chromosome 17 (DFTP-17), une dystrophie myotonique comme la maladie de Steinert, etc. [4, 5] chez des personnes âgées au niveau de la région hippocampique. Les agrégats de protéines tau peuvent se présenter sous différentes formes (les corps de Pick de la maladie de Pick, les touffes gliales de la paralysie supranucléaire progressive et les plaques gliales de la dégénérescence cortico-basale), et s’accumuler dans différents types cellulaires (neurones, astrocytes, oligodendrocytes). À chaque situation pathologique correspond une distribution laminaire et régionale spécifique d’agrégats de protéines tau. Cette observation indique que différentes sous-populations cellulaires sont affectées dans chaque maladie neurodégénérative. Si la dégénérescence neurofibrillaire n’est pas spécifique de la maladie d’Alzheimer, il est clair que sa distribution cérébrale reflète un processus étiopathogénique particulier.
Les protéines tau
Les protéines tau sont les constituants majeurs des filaments pathologiques intraneuronaux observés lors de la dégénérescence neurofibrillaire. Ces protéines tau sont alors agrégées et anormalement phosphorylées. Leur caractérisation biochimique par la technique des immuno-empreintes révèle la présence d’un triplet majeur de protéines phosphorylées (tau 60, 64 et 69), également appelé A68 ou tau-PHF.
Les protéines tau normales
Les protéines tau appartiennent à la famille des MAP (microtubule-associated proteins). Elles sont principalement neuronales et jouent un rôle dans la polymérisation des microtubules.
Dans le cerveau humain adulte, il existe six isoformes de protéines tau engendrées par épissage alternatif des exons 2, 3 et 10 d’un transcrit primaire d’un gène unique situé sur le chromosome 17 (Figure 1). Il faut noter que l’expression des protéines tau est réglée au cours du développement. Ainsi, une seule isoforme est présente à la naissance et elle ne comporte pas d’insertions codées par les exons 2, 3 ou 10, il s’agit de l’isoforme fœtale. Après la naissance, les autres isoformes vont apparaître au cours du développement. La longueur de leurs séquences varie de 352 à 441 acides aminés. Dans un gel de polyacrylamide en présence de sodium dodécyl sulfate (SDS-PAGE), les protéines tau normales migrent à une distance qui correspond à des protéines de 45 à 65 kDa.
 |
Figure 1. Les six isoformes de protéines tau. Chez l’homme, le gène des protéines tau est localisé sur le chromosome 17, à la position 17q21. Le transcrit primaire contient 16 exons. Dans le cerveau, les exons 4A, 6 et 8 ne sont pas traduits. Les exons 2, 3 et 10 sont épissés de manière alternative et sont spécifiques du tissu cérébral adulte. L’épissage alternatif de ces trois exons produit six combinaisons possibles (2-3-10-[352 aa] ; 2+3-10- [381 aa] ; 2+3+10- [410 aa] ; 2-3-10+ [383 aa] ; 2+3-10+ [412 aa] ; 2+3+10+ [441 aa]). Il y a donc six isoformes de protéines tau dans le cerveau adulte. Une seule isoforme est présente à la naissance et ne comporte pas d’insertions codées par les exons 2, 3 ou 10, il s’agit de l’isoforme foetale (352 aa). Après la naissance, les autres isoformes vont apparaître au cours du développement. Il y a donc trois isoformes à trois domaines de liaison aux microtubules (R1, R3 et R4) et trois isoformes à quatre domaines (R1 à R4). R1-R4 schématisent les quatre domaines de liaison aux microtubules. L’exon 10 code pour R2. |
Les protéines tau sont des phosphoprotéines impliquées dans la dynamique de polymérisation et de stabilisation des microtubules. Cette régulation relève de la structure primaire de la protéine tau qui correspond à trois ou quatre domaines d’interaction protéines tau-dimères de tubuline. Elle est également modulée par une modification post-traductionnelle, la phosphorylation. En premier lieu, les interactions avec les dimères de tubuline s’effectuent via le domaine carboxy-terminal des protéines tau. Ce dernier possède des séquences répétées qui se lient aux microtubules. Les trois isoformes de tau dépourvues de la séquence codée par l’exon 10 possèdent trois domaines de liaison aux microtubules (3R) et les trois isoformes comportant cette séquence possèdent quatre de ces régions répétitives (4R) (Figure 1). Dans un cerveau normal, la proportion d’isoformes 3R et 4R est identique. Cependant, l’interaction avec les microtubules est beaucoup plus forte pour les formes à 4 domaines, ce qui leur confère une meilleure stabilité. En second lieu, les protéines tau règlent également la stabilité des microtubules en fonction de leur état de phosphorylation. Il existe environ 80 résidus sérine et thréonine sur la protéine tau dont plus d’une vingtaine sont phosphorylés (Figure 2). La phosphorylation est donc la principale modification post-traductionnelle des protéines tau, surtout de part et d’autre des domaines de liaison aux microtubules. Une phosphorylation des protéines tau, en particulier dans la région riche en proline située en amont des motifs répétés, diminue leur affinité pour les microtubules, entraînant la dépolymérisation de ces derniers. De nombreuses sondes immunologiques, comme AD2, AT8, AT180 et AT270, reconnaissent spécifiquement ces sites phosphorylés (Figures 2 et 3). Par ailleurs, la phosphorylation de sites KXGS sur les résidus 262 et 356 (selon la numérotation de l’isoforme la plus longue), situés respectivement dans le premier et le quatrième domaine de liaison, est détectée grâce à l’anticorps 12E8. La phosphorylation de ces sites module également l’affinité des protéines tau pour ces microtubules (Figure 3) [6].
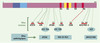 |
Figure 2. Phosphorylation des protéines tau et outils immunologiques. Il existe 80 sites potentiels de phosphorylation (Ser ou Thr) sur la protéine Tau. Certains sont des sites « normaux » de phosphorylation reconnus par des anticorps anti-tau dépendant de la phosphorylation (AD2, AT8, AT180, AT270 et 12E8). Cette phosphorylation règle la liaison de tau aux microtubules. Les sites reconnus par l’anticorps 12E8 sont cruciaux dans cette liaison de tau aux microtubules. Les sites « pathologiques » sont ceux de la phosphorylation anormale des protéines tau qui caractérise les tauopathies. Trois ont été identifiés et sont reconnus par les anticorps AT100, PHF-27/TG-3 et AP422/988. La numérotation des acides aminés reconnus est celle de l’isoforme la plus longue (441 acides aminés). |
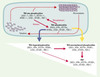 |
Figure 3. La phosphorylation normale et pathologique de tau. Dans un neurone, la dynamique des microtubules (équilibre tubuline-microtubules) est assurée par un équilibre entre les formes de protéines tau peu ou non phosphorylées et les formes phosphorylées. Les protéines tau présentent donc une immunoréactivité différente pour les anticorps décrits sur la Figure 2. Dans un neurone en dégénérescence neurofibrillaire, il y a hyperphosphorylation et/ou phosphorylation anormale des six isoformes de protéines tau et augmentation de l’immunoréactivité des protéines tau pour certains anticorps et/ou apparition de nouveaux épitopes (reconnus par exemple par les anticorps AT100 et 988). - : pas de reconnaissance par l’anticorps (pas de phosphorylation) ; -/+ : faible reconnaissance (faible phosphorylation) ; ± : reconnaissance (phosphorylation) et + : forte reconnaissance (phosphorylation importante). |
Les kinases impliquées dans la phosphorylation des protéines tau in vitro sont nombreuses. Parmi les plus communes, citons les PDPK (proline-directed protein kinases) dont les Cdk (kinases dépendantes Òdes cyclines), la GSK3β (glycogen synthase kinase 3) et la famille des MAP-kinases (ERK et SAPK) qui phosphorylent les résidus Ser et Thr suivis d’une proline. Parmi les kinases n’appartenant pas au groupe PDPK, les MARK (microtubule-affinity regulating kinases) , la phosphorylase K, la PKA (protéine kinase A), la PKC (protéine kinase C) et la tau-tubuline kinase peuvent aussi phosphoryler tau [7]. Il est aussi clairement établi qu’il existe un équilibre phosphorylation-déphosphorylation réglé par les phosphatases 1, 2A et 2B [8].
L’agrégation des protéines tau en PHF dans la maladie d’Alzheimer reste encore mystérieuse. L’ubiquitinylation et la glycation des protéines tau pourraient favoriser leur agrégation. Des co-facteurs tels que l’apolipoprotéine E, les glycosaminoglycanes et autres polyanions favoriseraient également l’agrégation des protéines tau en filaments [9].
Les protéines tau pathologiques de la maladie d’Alzheimer
Les protéines tau isolées des PHF sont anormalement phosphorylées (Figure 4). Ces protéines comportent des sites de phosphorylation ou des conformations qui leur sont spécifiques et qui sont révélés avec des anticorps tels que AP422/ 988, AT100 et TG3 (Figures 2, 3 et 4). Mais, la phosphorylation des protéines tau est incontestablement anormale dans la maladie d’Alzheimer, il reste à déterminer si elle résulte d’une activité kinase ou phosphatase anormale (Figures 3 et 4).
 |
Figure 4. Isoformes des protéines tau et mobilité électrophorétique : influence de la phosphorylation. Il est possible de visualiser les isoformes non phosphorylées, phosphorylées et anormalement phosphorylées de protéines tau par immuno-empreintes en utilisant les anticorps décrits dans les Figures 2 et 3. Les protéines tau non phosphorylées (par exemple, des protéines tau recombinantes) ne sont pas détectées avec des anti-tau dépendant de la phosphorylation (AD2 et AT100). Seul un anti-Tau indépendant de la phosphorylation (tau) permet de les visualiser avec une masse moléculaire apparente de 45 à 65 kDa. La phosphorylation normale des isoformes de protéines tau conduit à un ralentissement de leur mobilité électrophorétique qui se caractérise par une masse moléculaire apparente plus élevée de 60 à 74 kDa. Ces variants phosphorylés forment un triplet majeur de 60, 64 et 69 kDa avec un variant mineur à 74 kDa. Ils sont reconnus par un anticorps anti-tau et des anticorps anti-tau dépendant de la phosphorylation (AD2). En revanche, des anticorps spécifiques de la phosphorylation anormale (AT100) ne reconnaissent pas ces variants phosphorylés. Comme les variants normalement phosphorylés, les six variants de tau anormalement phosphorylés extraits de PHF (tau-PHF) apparaissent par immuno-empreintes sous la forme d’un triplet de 60, 64 et 69 kDa et d’une bande mineure à 74 kDa, et sont détectables par des sondes immunologiques spécifiques (AT100). Ils sont également reconnus par les autres anticorps (AD2 et tau). |
La majorité des études indique que, dans la maladie d’Alzheimer, les protéines tau ont une acidité plus importante, suggérant qu’elles comportent davantage de groupements phosphates. Certains sont spécifiques de résidus comme la Ser422. Celle-ci n’est phosphorylée que dans les protéines tau agrégées, mais pas dans les protéines tau normales ou fœtales (pour revue, voir [9]).
Quels sont les mécanismes qui peuvent expliquer cette phosphorylation anormale ? De nombreuses hypothèses ont été émises : neurotoxicité Aβ, perturbation de l’homéostasie du calcium, réactivation du cycle cellulaire, apoptose… Elles sont pour la plupart non exclusives ou même synergiques mais aucune n’est clairement démontrée. La mise au point de paradigmes expérimentaux permettant de modéliser cette phosphorylation anormale devrait aboutir à une meilleure compréhension des mécanismes moléculaires y conduisant.
Une analyse plus fine des variants de protéines tau dans la maladie d’Alzheimer a conduit à identifier la correspondance entre isoformes et variants phosphorylés de ces protéines. De plus, elle a permis l’identification d’un nouveau variant (tau 74) correspondant à l’hyperphosphorylation de l’isoforme la plus longue. Cette isoforme est également la moins exprimée. En effet, il faut noter que quatre isoformes de protéines tau sont dites majeures. En revanche, les deux isoformes comportant les séquences codées par les exons 2 et 3 (2+3+10- et 2+3+10+) sont présentes, mais en plus faible quantité. Ainsi, les six isoformes de protéines tau sont hyperphosphorylées dans la maladie d’Alzheimer et conduisent à une signature biochimique particulière, caractérisée en électrophorèse par un triplet majeur à 60 (tau 60), 64 (tau 64) et 69 (tau 69) kDa et un variant mineur à 74 kDa (tau 74) (Figures 4 et 5) [10].
 |
Figure 5. Caractérisation du profil électrophorétique des protéines tau anormalement phosphorylées dans les tauopathies. Les six variants anormalement phosphorylés de protéines tau qui forment majoritairement un triplet dans la maladie d’Alzheimer sont également détectés dans la trisomie 21, le syndrome de l’île de Guam et le syndrome parkinsonien post-encéphalitique. Au contraire, dans la paralysie supranucléaire progressive et la dégénérescence corticobasale, seules les isoformes de protéines tau anormalement phosphorylées à quatre domaines de liaison aux microtubules, s’agrègent et sont visualisées par immuno-empreintes comme un doublet majeur à 64 et 69 kDa avec une bande mineure à 74 kDa. Dans la maladie de Pick, les isoformes hyperphosphorylées à trois domaines de liaison aux microtubules s’agrègent en filaments et forment un doublet de 60 et 64 kDa avec un variant mineur de 69 kDa. Sur un schéma du cortex cérébral, il existe six couches dans l’isocortex. La distribution laminaire et régionale des lésions neuropathologiques d’agrégats de protéines tau (dégénérescence neurofibrillaire et corps de Pick) est différente pour une tauopathie donnée car les sous-populations neuronales qui dégénérent sont différentes. Ainsi, les différents profils électrophorétiques seraient liés à la dégénérescence de sous-populations neuronales différentes possédant un pool spécifique d’isoformes de protéines tau (E10+ : avec l’exon 10. E10- : sans l’exon 10 [5, 9, 16]). |
Les protéines tau des tauopathies
La notion de tauopathies, maladies liées à l’agrégation spécifique d’isoformes de protéines tau, était née. Elle repose sur le fait que les isoformes de protéines tau définissent des sous-populations neuronales spécifiques [5]. Dans le syndrome de l’île de Guam (→) et la maladie de Parkinson post-encéphalitique, il existe un triplet de protéines tau semblable à celui de la maladie d’Alzheimer (Figure 5) [11, 12]. Au contraire, la paralysie supranucléaire progressive (PSP) se caractérise par un autre profil électrophorétique correspondant à la présence d’un doublet de variants tau 64 et 69 et d’une forme mineure de 74 kDa. Ces variants sont également retrouvés dans la dégénérescence cortico-basale [13]. Ce profil particulier reflète l’agrégation sélective des isoformes de la protéine tau à quatre domaines de liaison aux microtubules (4R) (Figure 5) [14, 15].
(→) m/s 1987, n°8, p. 496
Dans la maladie de Pick, une démence fronto-temporale, un autre doublet de variants, tau 60 et 64, et une forme mineure, tau 69, sont détectés [13]. Les corps de Pick sont localisés principalement dans les couches II et VI de l’isocortex et les neurones granulaires du gyrus denté. Ces neurones ne contiennent pas les isoformes 10+ de protéines tau [16]. Or, seules les isoformes de protéines tau hyperphosphorylées dépourvues de la séquence codée par l’exon 10 (3R) présentent un tel profil électrophorétique [15, 17]. Il est donc clair que des isoformes ne comportant pas cette séquence s’agrègent au sein des corps de Pick (Figure 5). De plus, certains sites (KXGS), tels que les résidus Ser262 et 356, ne sont pas phosphorylés, suggérant l’absence ou l’inactivation des kinases impliquées dans cette phosphorylation, ou encore un problème d’accessibilité à ces sites [15, 16].
Le profil électrophorétique des protéines tau permet donc de différencier certaines maladies neurodégénératives. Ainsi, les différences biochimiques observées dans les maladies neurodégénératives seraient liées à la présence de différentes combinaisons d’isoformes de protéines tau (3R et 4R) dans des sous-populations neuronales vulnérables (Figure 5) [9].
Génétique des tauopathies
La découverte de formes familiales de démences frontotemporales résultant de mutations sur le gène TAU a permis de mieux comprendre comment les protéines tau pouvaient participer au mécanisme de dégénérescence neurofibrillaire. Il existe également des altérations dans l’expression et l’épissage de TAU dans d’autres tauopathies comme la dystrophie myotonique de Steinert [18] ou les démences fronto-temporales sans lésion histologique [19], mais elles ne seront pas développées dans cet article.
Les démences fronto-temporales avec syndrome parkinsonien liées au chromosome 17 (DFTP-17)
Historiquement, ces formes familiales hétérogènes ont été nommées en décrivant leurs signes cliniques caractéristiques (DDPAC : disinhibition-dementia-parkinsonism-amyotrophy complex ; FPDP : familial form of presenile dementia with psychosis ; HFTD : hereditary fronto-temporal dementia). Cependant, malgré une hétérogénéité des signes cliniques entre patients d’une même famille, et entre familles, certaines caractéristiques communes sont observées. Ainsi, les premiers signes apparaissent vers 40-50 ans et la progression de la maladie est généralement lente. Des troubles comportementaux caractéristiques d’une désinhibition frontale (comportement agressif ou obsessionnel) sont souvent les premiers symptômes observés. Les déficits cognitifs affectent les capacités de jugement, de planification et de raisonnement, sans perte de mémoire ou de l’orientation en début d’évolution. L’aggravation progressive sur plusieurs années peut conduire à une démence sévère. Les déficits moteurs, souvent peu visibles dans les premiers stades, se traduisent par une bradykinésie avec rigidité axiale et des membres, puis une instabilité posturale caractéristique du syndrome parkinsonien. Cependant, celui-ci n’est pas constant.
Au plan neuropathologique, les DFTP-17 regroupent des familles qui toutes présentent une gliose sous-corticale progressive ou une dégénérescence pallido-ponto-nigrale. Il est maintenant établi que l’atrophie corticale fronto-temporale est une constante des différentes formes de DFTP-17, souvent accompagnée d’une atrophie de structures sous-corticales comme la substance noire ou les noyaux de la base. La perte neuronale est importante, essentiellement dans les couches corticales superficielles et la substance noire. Un phénomène de gliose affecte la substance grise et la substance blanche. Des inclusions argentophiles positives pour tau sont observées, selon les familles, soit exclusivement dans les neurones, soit dans les neurones et dans les cellules gliales du néocortex et de certaines structures sous-corticales. Enfin, il n’y a, en général, pas de dépôts amyloïdes.
Actuellement, une vingtaine de mutations différentes de TAU ont été mises en évidence chez les patients atteints d’une DFTP-17 (pour revues, voir [4, 5, 9]). Elles ségrègent toujours avec la maladie, et ne sont jamais retrouvées chez les sujets témoins, soulignant l’implication directe des protéines tau mutées dans le processus pathogénique. Les mutations sont localisées soit dans la partie codante du gène des protéines tau, soit dans la séquence intronique qui suit l’exon 10. Les mutations exoniques sont situées dans la séquence codant pour le domaine de liaison aux microtubules, ou à proximité immédiate (Tableau I) [4, 5, 9, 20–22]. Les cinq mutations introniques (+3, +12, +13, +14, +16) sont proches du site d’épissage de l’exon 10. Une sixième mutation intronique ayant un effet potentiellement pathogène a été récemment décrite en position +33 (Tableau I) [4, 5, 9]. Une classification de ces mutations peut être établie sur la base de leurs conséquences fonctionnelles. Un premier groupe, comprenant la plupart des mutations non-sens situées dans le domaine de liaison aux microtubules, a pour effet de modifier la capacité de liaison des protéines tau aux microtubules [4, 5, 9]. Les mutations du deuxième groupe, correspondant aux mutations introniques plus quelques mutations exoniques, ont pour effet de modifier l’épissage de l’exon 10, et donc de changer la proportion des isoformes tau-4R par rapport aux isoformes tau-3R [4, 5, 9].
DFTP-17 : effets des mutationstau. SpE10 : épissage de l’exon 10 ; MT-tau : affinité de tau pour les microtu-bules ; I10 : intron 10 ; R1, R2, R3, R4 : identification des quatre domaines de liaison aux microtubules ; IR1-2, IR2-3, IR3-4 : trois domaines séparant les domaines de liaison aux microtubules ; + : augmenté ; - : diminué ; = : inchangé ; ? : indéterminé. PSP : paralysie supranucléaire progressive ; DCB : dégénérescence cortico-basale ; E : exon ; I : intron.
L’étude in vitro de l’effet des mutations non-sens du premier groupe montre que les protéines tau mutées se lient moins aux microtubules et ne permettent pas une bonne polymérisation de la tubuline en microtubules. Cela a été récemment vérifié in situ, montrant que la plupart des mutations non-sens, à l’exception de la mutation R406W, ne modifient pas la phosphorylation des protéines tau mais bien leur liaison aux microtubules [23]. Lorsque la mutation est située dans la région exonique commune aux six isoformes, et en dehors de l’exon 10 (G272V, V337M), les six isoformes perdent leur fonctionnalité. Elles s’agrègent alors sous forme de PHF (périodicité de 80 nm) et de filaments droits identiques à ceux observés au cours de la maladie d’Alzheimer. Seuls les neurones présentent alors ce type d’inclusions. L’analyse biochimique montre un profil électrophorétique de type « triplet MA » (MA pour maladie d’Alzheimer) tau 60, 64, 69 [4, 5, 9]. Lorsque la mutation non-sens est localisée dans l’exon 10 (P301L, P301S), seules les isoformes tau-4R sont affectées et perdent leur capacité de liaison aux microtubules. Elles s’agrègent alors pour former des filaments torsadés au sein des neurones et des cellules gliales. Le profil électrophorétique est également un doublet « type PSP » tau 64, 69 [4, 5, 9]. Dans le cas des mutations K257T, S320Y et G389R, des lésions neuropathologiques de type corps de Pick sont observées. De plus, le profil électrophorétique est du type « doublet Pick » tau 60, 64. Il est cependant intéressant de noter que, pour la mutation G389R, et contrairement à ce qui est décrit dans la maladie de Pick et les mutations K257T et S320Y, les corps de Pick sont immunoréactifs vis-à-vis de l’anticorps 12E8 spécifique de l’épitope phosphorylé des sites KXGS (Ser262/Ser356) [4, 5, 9]. Une autre mutation, K369I, est caractérisée par la présence de corps de Pick, mais son profil électrophorétique est de type Alzheimer (tau 60, 64 et 69). Il s’agit donc d’un phénotype purement DFTP-17 [21].
Les mutations introniques (+3, +12, +13, +14, +16) provoquent la désorganisation d’une structure en épingle à cheveux qui stabilise la région du transcrit primaire comportant le site d’épissage de l’exon 10. Elles facilitent ainsi l’utilisation de ce site, et sont donc responsables d’une formation accrue d’ARNm 10+ et donc d’une quantité plus importante d’isoformes tau-4R, la quantité totale de protéines tau solubles étant stable. Certaines mutations exoniques vont également modifier l’épissage de l’exon 10 en créant un élément activateur du site d’épissage ou en détruisant un élément répresseur (Tableau I). Dans ces deux dernières situations, la conséquence est la même que celle décrite précédemment pour les mutations introniques. Chez les patients possédant ce type de mutations, seules les isoformes tau-4R, traduites en plus grande quantité, s’agrègent sous forme de filaments torsadés présentant une périodicité irrégulière de 90-130 nm. Ces inclusions sont observées à la fois dans les neurones et dans les cellules gliales. La caractérisation biochimique des protéines tau met en évidence un profil électrophorétique de type « doublet PSP », c’est-à-dire un doublet majeur tau 64, de 69 kDa, avec une composante mineure à 72-74 kDa [4, 5, 9].
La mutation par délétion DK280 est un cas particulier d’un point de vue fonctionnel, puisqu’elle réduit l’affinité des protéines tau pour les microtubules, tout en modifiant la proportion d’isoformes tau-4R/tau-3R. Le mécanisme moléculaire produisant cet effet n’est pas déterminé, ni la caractérisation biochimique des protéines tau [4, 5, 9].
Génétique de la pathologie tau de la maladie d’Alzheimer
De nombreuses études ont suggéré une association entre des polymorphismes du gène codant pour la protéine tau et la maladie d’Alzheimer. Ces études étaient souvent contradictoires. Une récente méta-analyse de tous ces travaux a été réalisée et ne montre aucun lien entre la maladie d’Alzheimer à début tardif et les polymorphismes de la protéine tau [24].
Modélisation de la dégénérescence neurofibrillaire
Les souris transgéniques pour le gène TAU humain,avec trois ou quatre domaines de liaison aux microtubules, ne développent pas de dégénérescence neurofibrillaire. Cependant, il y a une accumulation de protéines tau phosphorylées dans le domaine somato-dendritique des neurones et présence d’amas sphéroïdes dans les axones, mais pas de filaments ou de phosphorylations anormales des protéines tau [25, 26]. En revanche, si le gène murin est invalidé, les souris transgéniques pour le gène TAU humain présentent une dégénérescence neurofibrillaire. Les modélisations cellulaires et animales ont permis de montrer que les mutations des DFTP-17 affectaient non seulement la dynamique des microtubules [27] mais étaient aussi capables d’entraîner une dégénérescence neurofibrillaire. Ainsi, les souris transgéniques pour la protéine tau humaine mutée en P301L avec expression neuronale montrent des dégénérescences neurofibrillaires avec une phosphorylation anormale des protéines tau [28]. Ces anomalies sont-elles suffisantes pour expliquer la pathologie humaine ? Des études complémentaires sont nécessaires pour répondre à cette question.
La protéine tau dans la dégénérescence neurofibrillaire
On peut conclure de toutes ces observations sur les maladies neurodégénératives sporadiques et familiales, regroupées sous le nom de tauopathies, que les protéines tau peuvent être considérées comme des marqueurs et des acteurs du processus de dégénérescence neurofibrillaire. L’expression d’isoformes de protéines tau et de certaines kinases permet de définir un phénotype cellulaire des sous-populations neuronales vulnérables à une pathologie neurodégénérative. Par ailleurs, pour certaines formes familiales de tauopathies, il existe une composante génétique qui va modifier l’épissage alternatif de TAU et, par conséquent, la proportion relative d’isoformes de tau exprimées. Cette modification est suffisante pour conduire à une dégénérescence neurofibrillaire.
Dans la plupart des cas, l’agrégation et la phosphorylation anormale des protéines tau caractérisent le processus de dégénérescence neurofibrillaire. On notera qu’il existe également des maladies ayant une expression anormale des isoformes tau ou une diminution de l’expression des protéines tau normales.
L’ensemble de ces données suggère que l’absence d’interactions entre protéines tau et microtubules conduit à une dégénérescence neuronale qui se caractérise par une phosphorylation anormale des protéines tau et leur agrégation en filaments.
Les relations entre les pathologies APP et les protéines tau
Vers une compréhension de la maladie d’Alzheimer
Il est maintenant admis que l’amyloïdogenèse et la pathologie tau sont les deux processus qui caractérisent la pathologie Alzheimer. Il reste pourtant à déterminer leur importance et leur rôle respectif dans l’induction du phénomène de dégénérescence ou dans sa propagation.
Déterminer les relations entre les pathologies Aβ et tau semble actuellement la question primordiale pour la compréhension de l’étiologie de la maladie d’Alzheimer.
Il est très difficile d’établir une cinétique d’apparition des dépôts amyloïdes dans la maladie d’Alzheimer. Selon Braak et Braak [29], trois stades sont à distinguer selon la gravité de la surcharge amyloïde, mais il n’y a pas de consensus sur une distribution régionale et laminaire de ces dépôts spécifique de la maladie d’Alzheimer. À l’inverse, une séquence d’apparitions spatio-temporelle dans la dégénérescence neurofibrillaire est tout à fait caractéristique et seules certaines régions corticales bien définies sont touchées au début de la maladie. La pathologie tau localisée à la formation hippocampique peut être attribuée au vieillissement cérébral « normal » ou à une pathologie tau qui frappe tardivement et électivement cette région cérébrale. La dégénérescence neurofibrillaire suit des voies établies [30, 31]. L’atteinte du cortex temporal est un phénomène précoce dans la maladie et se traduit par de légers signes cliniques. Il ne s’agit donc pas d’un vieillissement cérébral normal [32]. Une analyse biochimique de 130 cas de sujets non déments et déments permet d’établir la séquence suivante : la dégénérescence neurofibrillaire commence dans la région hippocampique (cortex transentorhinal, entorhinal, puis hippocampe) et s’étend séquentiellement aux régions temporales (aires de Brodmann 38, 20, 21). Puis elle touche les régions associatives polymodales (aires de Brodmann 22 [temporale], 39 [pariétale] et 9 [frontale]). Dans les cas les plus sévères, elle peut être retrouvée dans des régions sensitives primaires (aires 4 [motrice] et 17 [visuelle]). Ces résultats confirment donc qu’il existe des sous-populations neuronales vulnérables et que la pathologie tau progresse par les voies de connexions nerveuses cortico-corticales [33]. Ils corroborent les résultats obtenus dans les autres tauopathies dans lesquelles la distribution laminaire et régionale des agrégats de protéines tau est différente selon les pathologies. Ces différences sont vraisemblablement liées à la vulnérabilité différentielle de sous-populations neuronales particulières en fonction de l’agent étiologique et/ou du sous-type de pathologies tau.
La dégénérescence neurofibrillaire de sous-populations neuronales particulières dans la maladie d’Alzheimer est vraisemblablement liée à un dysfonctionnement du métabolisme de l’APP (amyloid precursor protein), le précurseur du peptide Aβ, qui rend vulnérables certaines sous-populations neuronales [34]. Ce dysfonctionnement de l’APP pourrait également contribuer à l’extension de la pathologie tau dans les aires cérébrales [35].
Vers une modélisation de la maladie d’Alzheimer
La modélisation de la synergie des pathologies tau et Aβ est difficile à appréhender. Deux études récentes ont renforcé l’idée de cette synergie. Elle utilisent toutes deux des souris transgéniques pour la protéine tau humaine avec la mutation P301L, qui présentent une dégénérescence neurofibrillaire. La première étude a consisté à croiser ces souris avec des souris transgéniques pour l’APP comportant la double mutation suédoise (Lys670→Asn, Met671→Leu). Les souris double transgéniques présentaient une dégénérescence neurofibrillaire beaucoup plus importante, suggérant que l’APP muté ou le peptide Aβ potentialise ce processus pathologique [36]. Dans l’autre étude, le peptide Aβ(1-42) agrégé a été directement injecté dans l’hippocampe de souris transgéniques tau P301L. Une augmentation de la dégénérescence neurofibrillaire est observée dans certaines régions de projection de l’hippocampe telles que l’amygdale [37]. Ces deux études démontrent clairement les interactions entre la pathologie tau et APP. De plus, elles suggèrent que la pathologie amyloïde potentialise la pathologie tau pré-existante, mais ne l’induit pas. Ces études sont en accord avec ce qui est observé dans le cerveau humain [35, 38].
Conclusions
La compréhension de la maladie d’Alzheimer a bénéficié à la fois de la génétique, de la neuropathologie et de la biochimie des maladies neurodégénératives. Le diagnostic biologique de la maladie d’Alzheimer pourrait également bénéficier de ces recherches car une quantification précise des protéines tau et du peptide Aβ dans le liquide céphalo-rachidien serait aujourd’hui techniquement envisageable. Mais l’aspect éthique de la question reste à traiter et un tel diagnostic devrait sans doute être amélioré en détectant plus précisément certaines isoformes de protéines tau. En tout état de cause, on peut désormais répondre à la question posée dans le titre de cet article : la maladie d’Alzheimer est une tauopathie dont la pathologie est potentialisée par le dysfonctionnement de l’APP.
Remerciements
Ce travail a bénéficié d’un soutien du Conseil Régional Nord-Pasde-Calais dans le cadre de la Génopole de Lille, de l’Inserm et du Cnrs. Nous remercions également tous nos collègues scientifiques et cliniciens pour leurs discussions enrichissantes.
Références
- Alzheimer A. Über eine eigenartige erkrankung der hirnrinde. Allg Z Psychiat 1907; 64 : 146–8. [Google Scholar]
- Glenner GG, Wong CW. Alzheimer’s disease : initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984; 120 : 885–90. [Google Scholar]
- Brion JP, Passareiro H, Nunez J, Flament-Durand J. Immunological determinants of tau proteins are present in neurofibrillary tangles of Alzheimer’s disease. Arch Biol (Brux) 1985; 95 : 229–35. [Google Scholar]
- Lee VMY, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Ann Rev Neurosci 2001; 24 : 1121–59. [Google Scholar]
- Delacourte A, Buée L. Tau pathology : a marker of neurodegenerative disorders. Curr Opin Neurol 2000; 13 : 371–6. [Google Scholar]
- Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules. Neuron 1993; 11 : 153–63. [Google Scholar]
- Lovestone S, Reynolds CH. The phosphorylation of tau : a critical stage in neurodevelopment and neurodegenerative processes. Neuroscience 1997; 78 : 309–24. [Google Scholar]
- Trojanowski JQ, Lee VMY. Phosphorylation of paired helical filament tau in Alzheimer’s disease neurofibrillary lesions: focusing on phosphatases. FASEB J 1995; 9 : 1570–6. [Google Scholar]
- Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein iso-forms, phosphorylation and role in neurodegenerative disorders. Brain Res Rev 2000; 33 : 95–130. [Google Scholar]
- Sergeant N, David J P, Goedert M, et al. Two-dimensional characterization of paired helical filament-tau from Alzheimer’s disease. J Neurochem 1997; 69 : 834–44. [Google Scholar]
- Buée-Scherrer V, Buée L, Leveugle B, et al. Pathological tau proteins in postencephalitic parkinsonism : comparison with Alzheimer’s disease and neurodegenerative disorders. Ann Neurol 1997; 42 : 356–9. [Google Scholar]
- Buée-Scherrer V, Buée L, Hof PR, et al. Neurofibrillary degenera-tion in amyotrophic lateral sclerosis/ parkinsonismdementia complex of Guam. Immunochemical characte-rization of tau proteins. Am J Pathol 1995; 146 : 924–32. [Google Scholar]
- Buée-Scherrer V, Hof PR, Buée L, et al. Hyperphosphorylated tau proteins differentiate corticobasal degeneration and Pick’s disease. Acta Neuropathol 1996; 91 : 351–9. [Google Scholar]
- Sergeant N, Wattez A, Delacourte A. Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusely « exon 10 » isoforms. J Neurochem 1999; 72 : 1243–9. [Google Scholar]
- Mailliot C, Sergeant N, Bussière T, Caillet-Boudin ML, Delacourte A, Buée L. Phosphorylation of specific sets of tau isoforms reflects different neurofibrillary degeneration processes. FEBS Lett 1998; 433 : 201–4. [Google Scholar]
- Delacourte A, Sergeant N, Wattez A, Gauvreau D, Robitaille Y. Vulnerable neuronal subsets in Alzheimer’s disease and Pick’s disease are distinguished by their tau iso-form distribution and phosphorylation. Ann Neurol 1998; 43 : 193–204. [Google Scholar]
- Sergeant N, David JP, Lefranc D, Vermersch P, Wattez A, Delacourte A. Different distribution of phosphorylated tau protein isoforms in Alzheimer’s and Pick’s diseases. FEBS Lett 1997; 412 : 578–82. [Google Scholar]
- Sergeant N, Sablonnière B, Schraen-Maschke S, et al. Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum Mol Genet 2001; 10 : 2143–55. [Google Scholar]
- Zhukareva V, Vogelsberg-Ragaglia V, Van Deerlim V, et al. Loss of brain tau defines novel sporadic and familial tauopathies with frontotemporal dementia. Ann Neurol 2001; 49 : 165–75. [Google Scholar]
- Iseki E, Matsumura T, Marui W, et al. Familial frontotemporal dementia and parkinsonism with a novel mutation N296H mutation in exon 10 of the tau gene and a widespread tau accumulation in the glial cells. Acta Neuropathol 2001; 102 : 285–92. [Google Scholar]
- Neumann M, Schultz-Schaeffer W, Crowther RA, et al. Pick’s disease associated with the novel Tau gene mutation K369I. Ann Neurol 2001; 50 : 503–13. [Google Scholar]
- Pastor P, Pastor E, Carnero C, et al. Familial atypical progressive supranuclear palsy associated with homozigosity for the delN296 mutation in the tau gene. Ann Neurol 2001; 49 : 263–7. [Google Scholar]
- Delobel P, Flament S, Hamdane M, et al. Functional characterization of FTDP-17 tau gene mutations through their effects on Xenopus oocyte maturation. J Biol Chem 2002; 277 : 9199–205. [Google Scholar]
- Russ C, Powell JF, Zhao J, et al. The microtubule associated protein Tau gene and Alzheimer’s disease - an association study and meta-analysis. Neurosci Lett 2001; 314 : 92–6. [Google Scholar]
- Götz J, Probst A, Spillantini MG, et al. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J 1995; 14 : 1304–13. [Google Scholar]
- Brion JP, Tremp G, Octave JN. Transgenic expression of the shortest human tau affects its compartmentalization and its phosphorylation as in the pretangle stage of Alzheimer’s disease. Am J Pathol 1999; 154 : 255–70. [Google Scholar]
- Götz J. Tau and transgenic animal models. Brain Res Rev 2001; 35 : 266–86. [Google Scholar]
- Lewis J, McGowan E, Rockwood J, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 2000; 25 : 402–5. [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82 : 239–59. [Google Scholar]
- Fewster PH, Griffin-Brooks S, MacGregor J, Ojalvo-Rose E, Ball MJ. A topographical pathway by which histopathological lesions disseminate through the brain of patients with Alzheimer’s disease. Dementia 1991; 2 : 121–32. [Google Scholar]
- Duyckaerts C, Colle MA, Dessi F, Grignon Y, Piette F, Hauw JJ. The progression of the lesions in Alzheimer disease: insights from a prospective clinicopathological study. J Neural Transm 1998; 53 (suppl) : 119–26. [Google Scholar]
- Delacourte A, Buée L, David JP, et al. Lack of continuum between cerebral aging and Alzheimer’s disease as revealed by PHF-tau and Aβ biochemistry. Alzh Rep 1998; 1 : 101–10. [Google Scholar]
- Delacourte A, David JP, Sergeant N, et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology 1999; 52 : 1158–65. [Google Scholar]
- Delacourte A. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology 2000; 54 : 538. [Google Scholar]
- Delacourte A, Sergeant N, Champain D, et al. The biochemical spreading of Tau and amyloid β precursor protein pathologies in aging and sporadic Alzheimer’s disease. Brain Aging 2001; 1 : 33–42. [Google Scholar]
- Lewis J, Dickson DW, Lin WL, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001; 293 : 1487–91. [Google Scholar]
- Götz J, Chen F, Van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 2001; 293 : 1491–5. [Google Scholar]
- Sergeant N, David JP, Champain D, et al. Progressive decrease of amyloid precursor protein carboxy-terminal fragments (APP-CTFs), associated with tau pathology stages, in Alzheimer’s disease. J Neurochem 2002; 81 : 663–72. [Google Scholar]
Liste des tableaux
DFTP-17 : effets des mutationstau. SpE10 : épissage de l’exon 10 ; MT-tau : affinité de tau pour les microtu-bules ; I10 : intron 10 ; R1, R2, R3, R4 : identification des quatre domaines de liaison aux microtubules ; IR1-2, IR2-3, IR3-4 : trois domaines séparant les domaines de liaison aux microtubules ; + : augmenté ; - : diminué ; = : inchangé ; ? : indéterminé. PSP : paralysie supranucléaire progressive ; DCB : dégénérescence cortico-basale ; E : exon ; I : intron.
Liste des figures
|
Figure 1. Les six isoformes de protéines tau. Chez l’homme, le gène des protéines tau est localisé sur le chromosome 17, à la position 17q21. Le transcrit primaire contient 16 exons. Dans le cerveau, les exons 4A, 6 et 8 ne sont pas traduits. Les exons 2, 3 et 10 sont épissés de manière alternative et sont spécifiques du tissu cérébral adulte. L’épissage alternatif de ces trois exons produit six combinaisons possibles (2-3-10-[352 aa] ; 2+3-10- [381 aa] ; 2+3+10- [410 aa] ; 2-3-10+ [383 aa] ; 2+3-10+ [412 aa] ; 2+3+10+ [441 aa]). Il y a donc six isoformes de protéines tau dans le cerveau adulte. Une seule isoforme est présente à la naissance et ne comporte pas d’insertions codées par les exons 2, 3 ou 10, il s’agit de l’isoforme foetale (352 aa). Après la naissance, les autres isoformes vont apparaître au cours du développement. Il y a donc trois isoformes à trois domaines de liaison aux microtubules (R1, R3 et R4) et trois isoformes à quatre domaines (R1 à R4). R1-R4 schématisent les quatre domaines de liaison aux microtubules. L’exon 10 code pour R2. |
| Dans le texte | |
|
Figure 2. Phosphorylation des protéines tau et outils immunologiques. Il existe 80 sites potentiels de phosphorylation (Ser ou Thr) sur la protéine Tau. Certains sont des sites « normaux » de phosphorylation reconnus par des anticorps anti-tau dépendant de la phosphorylation (AD2, AT8, AT180, AT270 et 12E8). Cette phosphorylation règle la liaison de tau aux microtubules. Les sites reconnus par l’anticorps 12E8 sont cruciaux dans cette liaison de tau aux microtubules. Les sites « pathologiques » sont ceux de la phosphorylation anormale des protéines tau qui caractérise les tauopathies. Trois ont été identifiés et sont reconnus par les anticorps AT100, PHF-27/TG-3 et AP422/988. La numérotation des acides aminés reconnus est celle de l’isoforme la plus longue (441 acides aminés). |
| Dans le texte | |
|
Figure 3. La phosphorylation normale et pathologique de tau. Dans un neurone, la dynamique des microtubules (équilibre tubuline-microtubules) est assurée par un équilibre entre les formes de protéines tau peu ou non phosphorylées et les formes phosphorylées. Les protéines tau présentent donc une immunoréactivité différente pour les anticorps décrits sur la Figure 2. Dans un neurone en dégénérescence neurofibrillaire, il y a hyperphosphorylation et/ou phosphorylation anormale des six isoformes de protéines tau et augmentation de l’immunoréactivité des protéines tau pour certains anticorps et/ou apparition de nouveaux épitopes (reconnus par exemple par les anticorps AT100 et 988). - : pas de reconnaissance par l’anticorps (pas de phosphorylation) ; -/+ : faible reconnaissance (faible phosphorylation) ; ± : reconnaissance (phosphorylation) et + : forte reconnaissance (phosphorylation importante). |
| Dans le texte | |
|
Figure 4. Isoformes des protéines tau et mobilité électrophorétique : influence de la phosphorylation. Il est possible de visualiser les isoformes non phosphorylées, phosphorylées et anormalement phosphorylées de protéines tau par immuno-empreintes en utilisant les anticorps décrits dans les Figures 2 et 3. Les protéines tau non phosphorylées (par exemple, des protéines tau recombinantes) ne sont pas détectées avec des anti-tau dépendant de la phosphorylation (AD2 et AT100). Seul un anti-Tau indépendant de la phosphorylation (tau) permet de les visualiser avec une masse moléculaire apparente de 45 à 65 kDa. La phosphorylation normale des isoformes de protéines tau conduit à un ralentissement de leur mobilité électrophorétique qui se caractérise par une masse moléculaire apparente plus élevée de 60 à 74 kDa. Ces variants phosphorylés forment un triplet majeur de 60, 64 et 69 kDa avec un variant mineur à 74 kDa. Ils sont reconnus par un anticorps anti-tau et des anticorps anti-tau dépendant de la phosphorylation (AD2). En revanche, des anticorps spécifiques de la phosphorylation anormale (AT100) ne reconnaissent pas ces variants phosphorylés. Comme les variants normalement phosphorylés, les six variants de tau anormalement phosphorylés extraits de PHF (tau-PHF) apparaissent par immuno-empreintes sous la forme d’un triplet de 60, 64 et 69 kDa et d’une bande mineure à 74 kDa, et sont détectables par des sondes immunologiques spécifiques (AT100). Ils sont également reconnus par les autres anticorps (AD2 et tau). |
| Dans le texte | |
|
Figure 5. Caractérisation du profil électrophorétique des protéines tau anormalement phosphorylées dans les tauopathies. Les six variants anormalement phosphorylés de protéines tau qui forment majoritairement un triplet dans la maladie d’Alzheimer sont également détectés dans la trisomie 21, le syndrome de l’île de Guam et le syndrome parkinsonien post-encéphalitique. Au contraire, dans la paralysie supranucléaire progressive et la dégénérescence corticobasale, seules les isoformes de protéines tau anormalement phosphorylées à quatre domaines de liaison aux microtubules, s’agrègent et sont visualisées par immuno-empreintes comme un doublet majeur à 64 et 69 kDa avec une bande mineure à 74 kDa. Dans la maladie de Pick, les isoformes hyperphosphorylées à trois domaines de liaison aux microtubules s’agrègent en filaments et forment un doublet de 60 et 64 kDa avec un variant mineur de 69 kDa. Sur un schéma du cortex cérébral, il existe six couches dans l’isocortex. La distribution laminaire et régionale des lésions neuropathologiques d’agrégats de protéines tau (dégénérescence neurofibrillaire et corps de Pick) est différente pour une tauopathie donnée car les sous-populations neuronales qui dégénérent sont différentes. Ainsi, les différents profils électrophorétiques seraient liés à la dégénérescence de sous-populations neuronales différentes possédant un pool spécifique d’isoformes de protéines tau (E10+ : avec l’exon 10. E10- : sans l’exon 10 [5, 9, 16]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.