")
")
| Issue |
Med Sci (Paris)
Volume 18, Number 6-7, Juin–Juillet 2002
|
|
|---|---|---|
| Page(s) | 717 - 724 | |
| Section | M/S Revues : Articles de Synthèse | |
| DOI | https://doi.org/10.1051/medsci/20021867717 | |
| Published online | 15 juin 2002 | |
Métabolisme du précurseur du peptide amyloïde et présénilines
Amyloid peptide precursor’metabolism and presenilins
1
IPMC du Cnrs, UMR 6097, 660, route des Lucioles, 06560 Valbonne, France
2
Inserm EPI 9906, 76183 Rouen, France
3
Laboratoire GlaxoSmithKline, 78163 Marly-le-Roi, France
4
Mount Sinai School of Medicine, New York, NY 10029, États-Unis
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La production exacerbée de peptide amyloïde contribue vraisemblablement à l’étiologie de la maladie d’Alzheimer. En effet, ce peptide s’accumule, s’agrège et conduit aux dépôts protéiques -les plaques séniles - qui envahissent le cortex des patients atteints de maladie d’Alzheimer. Une des approches thérapeutiques théoriques visant à ralentir ou à bloquer l’apparition des plaques séniles consiste donc à cibler les enzymes qui sont responsables de la formation du peptide amyloïde. Celles-ci sont regroupées sous le terme générique de sécrétases. La libération du peptide amyloïde fait intervenir de manière séquentielle la β-sécrétase puis la y-sécrétase. Une autre activité enzymatique, l’α-sécrétase, a un rôle opposé et protecteur puisqu’elle « détruit » le peptide amyloïde. Cet article présente les voies métaboliques de la maturation physiopathologique de la (βAPP et les avancées récentes de la recherche concernant la nature des sécrétases impliquées dans la maladie d’Alzheimer.
Abstract
Alzheimers’ disease is characterized by extracellular deposits called senile plaques, that are mainly composed by the amyloid-β peptide (Aβ). This peptide derives from the sequential proteolysis of a precursor protein (βAPP) by β- and γ-secretases. Familial forms of Alzheimer’s disease are due to autosomal dominant mutations on the genes encoding βAPP and two homologous proteins called presenilins. This paper reviews the physiopathological processing of the βAPP, the mechanisms by which presenilins influence such pathways and their control by the proteasome, and recent advances concerning the potential nature of the secretases.
© 2002 médecine/sciences - Inserm / SRMS
La maladie d’Alzheimer représente l’un des problèmes majeurs de santé publique des pays développés. L’allongement de la durée de vie permet de prédire que ce syndrome lié à l’âge présentera une incidence encore accrue dans les années à venir. La grande majorité des cas de maladie d’Alzheimer est d’origine sporadique, ce qui, en l’état des connaissances, signifie que la symptomatologie de la maladie intervient après 60 ans sans identification d’un quelconque déterminisme génétique. Les formes familiales (FAD pour familial Alzheimer’s disease) se caractérisent par un début beaucoup plus précoce (certains cas ont été décrits chez des patients de moins de 25 ans), une évolution plus rapide et la présence de mutations autosomiques dominantes sur les gènes codant pour une des trois protéines identifiées, la βAPP (pour β-amyloid precursor protein) et les présénilines 1 et 2 (pour revue voir [1]). Qu’elles soient sporadiques ou génétiques, toutes les formes de maladie d’Alzheimer se caractérisent - au stade tardif, et d’un point de vue histopathologique - par des lésions appelées plaques séniles envahissant le cerveau. Les plaques séniles sont des dépôts protéiques extracellulaires qui évoluent d’un point de vue morphologique et biochimique. En effet, les premières lésions correspondent à des dépôts amorphes (plaques diffuses) présents dans les noyaux sous-corticaux et sont essentiellement composées d’un fragment protéique de 42 acides aminés (Aβ42). Ces lésions évoluent vers des dépôts plus compacts appelés plaques mûres (ou plaques séniles) composées principalement d’un peptide amyloïde de 40 résidus (Aβ40). Les plaques séniles envahissent notamment les aires corticales associatives. Ce sont sans doute ces atteintes qui conditionnent l’apparition des premiers symptômes (en général des plaintes mnésiques).
Les formes familiales et sporadiques présentent les mêmes stigmates anatomiques mais se distinguent par la cinétique de leur apparition. Il était donc plus que probable que l’accélération du processus neurodégénératif soit vraisemblablement dû, au moins en partie, à l’exacerbation de la production de peptide amyloïde. Cette hypothèse a été rapidement corroborée par des approches de biologie cellulaire qui ont permis d’établir que toutes les mutations responsables de formes avérées de maladie d’Alzheimer se traduisaient par une modification de la production cellulaire de peptide amyloïde [2]. Le fait que des protéines distinctes (βAPP et présénilines) - qui lorsqu’elles sont mutées conduisent toutes à la maladie - présentent comme dénominateur commun une modification de la production de peptide Aβ, est de toute évidence un argument fort en faveur du rôle central du peptide amyloïde dans l’étiologie de la maladie d’Alzheimer. Le corollaire de cette observation est qu’une approche théorique visant à ralentir le processus pathologique consiste à cibler et à inhiber les sécrétases responsables de la production de peptide Aβ. Nous étudierons dans cet article les diverses voies métaboliques de la βAPP, la nature des sécrétases impliquées et le rôle des présénilines dans la maturation physiopathologique de la βAPP.
Maturation physiopathologique de la βAPP
Préalable
La βAPP est une protéine transmembranaire de 695 à 770 acides aminés codée par un seul gène [3]. Le peptide β-amyloïde résulte de la coupure séquentielle de la (βAPP par la (ou les) β- puis γ-sécrétase(s) qui libèrent respectivement les extrémités N- et C-terminales du peptide Aβ(Figure 1). La γ-sécrétase présente la particularité d’intervenir dans un environnement hydrophobe théoriquement hostile à l’hydrolyse puisque l’extrémité C-terminale du peptide Aβ est « enfouie » dans la membrane (Figure 1). C’est la γ-sécrétase qui conditionne la nature du peptide amyloïde engendré. Cela n’est pas anecdotique puisque les propriétés physicochimiques des peptides Aβ40 et 42 sont sensiblement différentes, le peptide de 42 acides aminés étant beaucoup plus hydrophobe et susceptible de s’agréger [4]. Une coupure alternative engendrée par l’α-sécrétase intervient au milieu de la séquence Aβ et prévient ainsi sa formation. Cette coupure libère un produit sécrété,l’APPα, qui présente une activité cytotrophique et neuroprotectrice (pour revue, voir [5]). Cette coupure correspond à la voie physiologique de maturation de la βAPP alors que les attaques séquentielles de la βAPP par les β-et γ-sécrétases représentent la voie amyloïdogénique de maturation.
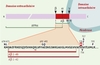 |
Figure 1. Structure schématique de la βAPP et mutations pathogènes. Structure transmembranaire de la βAPP ciblée par les β-et γ-sécrétases et engendrant le peptide Aβ. Une coupure alternative par l’α-sécrétase intervient dans la séquence Aβ et libère un fragment sécrété, l’APPα. Les principales mutations intervenant au niveau de la séquence Aβ sont représentées. |
Il faut souligner ici que la voie amyloïdogénique n’est pas systématiquement synonyme de pathologie. En effet, tout un chacun produit du peptide amyloïde. C’est le dérèglement de cette production qui est pathogène. Il peut se traduire soit par une augmentation de la concentration de peptide Aβ « normal », c’est-à-dire le peptide Aβ40, soit par l’altération du rapport des formes Aβ40/Aβ42. En effet, le peptide Aβ42 représente en général moins de 10 % de la totalité du peptide Aβ. En tout état de cause, les deux mécanismes conduisent vraisemblablement au passage d’un état « soluble » de ces peptides à un état agrégé, favorisant les dépôts et obérant vraisemblablement les processus de dégradation, d’où l’accumulation observée lors de la maladie.
L’équilibre entre la production et la dégradation du peptide Aβ se stabilise dans les conditions normales à un seuil en dessous duquel le peptide Aβ reste soluble. Le dérèglement de cet équilibre est sans doute très progressif et lent comme l’indiquent les débuts tardifs des formes sporadiques et l’évolution relativement lente du processus dégénératif. Comme cela est décrit au début de cet article, ce dérèglement s’accélère dans le cas des formes familiales. Une observation particulièrement intéressante réside dans le fait que toutes les mutations portées par la βAPP (elles sont maintenant au nombre de 17 – pour plus de détails, voir le site http://www.alzforum.org/) sont localisées soit au milieu de la séquence Aβ, soit bordent les extrémités N-et C-terminales du peptide (Figure 1). Dans les faits, cela se traduit par une production exacerbée de peptide Aβ (mutation suédoise N-terminale KM -> NL) ou par une coupure accrue au niveau 42 pour les mutations localisées près de l’extrémité C-terminale (Figure 1). Ce dernier phénotype est aussi observé pour les mutations portées par les présénilines (voir plus loin).
La voie physiologique de maturation de la βAPP
Il existe deux voies α-sécrétase distinctes : une voie sécrétoire constitutive et une voie de sécrétion réglée. Très brièvement, la voie réglée de production de l’APPα est sous le contrôle de la protéine kinase C (PKC) (pour revue, voir [6]). Elle conduit de manière concomitante à la diminution de la production de peptide Aβ, en accord avec l’hypothèse selon laquelle les productions de peptide Aβ et d’APPα ne seraient pas mutuellement exclusives. Plus récemment, nous avons établi que la voie α-sécrétase pouvait aussi être contrôlée par une activité de type protéine kinase A (PKA) dépendante de l’AMPc [7]. Les cibles de la PKA semblent différentes de celles de la PKC puisque la stimulation de la voie PKA entraîne aussi une augmentation de peptide Aβ, ce qui nous a conduits à proposer que la PKA ciblerait une protéine située en amont des α- et β/γ-sécrétases [7], qui pourrait être impliquée dans les mécanismes de bourgeonnement intervenant très tôt lors des processus sécrétoires.
L’α-sécrétase intervient vraisemblablement dans deux compartiments cellulaires distincts. Une coupure intervient dans les compartiments golgiens, c’est-à-dire assez tôt dans les voies sécrétoires (pour revue, voir [3]). Il apparaît clairement qu’une fraction de la βAPP échappe à cette protéolyse et est intégrée à la membrane plasmique où elle y subit l’attaque protéolytique (Figure 2). La nature des compartiments où la protéolyse s’effectue permettait de supposer, a priori, l’occurrence d’α-sécrétases distinctes puisque le pH des organites (acide dans le Golgi ou neutre au niveau de la membrane plasmique) soustend des propriétés physico-chimiques distinctes des enzymes intracellulaires et membranaires. Ce distingo empirique a été corroboré par l’identification des α-sécrétases membranaires. Celles-ci appartiennent au groupe des désintégrines - des métalloprotéases généralement impliquées dans la coupure de protéines trans-membranaires ou ancrées par une attache de type glycosylphospho-inositide [8]. Il apparaît qu’ADAM10 (A disintegrin and metalloprotease) contribue aux deux voies - constitutive [9, 10] et réglée - alors qu’ADAM17 (aussi appelée TACE pour tumor necrosis (α-converting enzyme) est essentiellement impliquée dans la voie réglée dépendante de la PKC [11] (→). L’α-sécrétase intracellulaire n’a pas été formellement identifiée. Nous avons toutefois établi qu’une protéase intracellulaire de la famille des pro-hormones convertases (PC7) contribue vraisemblablement à la voie α-sécrétase intracellulaire de la βAPP [10, 12] sans pouvoir définitivement écarter la possibilité que PC7 intervienne de manière indirecte sur la maturation d’une α-sécrétase intracellulaire.
(→) m/s 1999, n°1, p. 117
 |
Figure 2. Voie non amyloïdogénique de maturation de la βAPP. L a βAPP est coupée dans le réseau trans-golgien (TGN) par une α-sécrétase intracellulaire. Cette coupure libère un fragment, l’APPα, qui est engendré par la voie sécrétoire constitutive ou réglé par la protéine kinase C. Un pool de βAPP échappe à cette coupure intracellulaire et est intégré dans la membrane plasmique où une α-sécrétase membranaire engendre la même coupure. Cette voie de maturation de la βAPP est considérée comme non amyloïdogénique puisque les α-sécrétases interviennent au milieu du peptide amyloïde (Aβ, segment représenté en rouge), prévenant ainsi sa formation. |
La voie amyloïdogénique de maturation de la βAPP
On peut distinguer deux voies majeures de production de peptide Aβ : une voie sécrétoire constitutive et une voie faisant intervenir un processus d’endocytose (Figure 3). Le peptide Aβ est détectable très tôt dans la voie sécrétoire. En effet, le peptide Aβ, et notamment la forme longue Aβ42, est décelable dans le réticulum endoplasmique, alors que le peptide Aβ40 semble produit dans le compartiment golgien [13]. La distribution subcellulaire distincte des deux peptides n’est pas antinomique de l’hypothèse selon laquelle une seule γ-sécrétase interviendrait. En effet, il a été suggéré que la « spécificité » apparente du type de coupure serait en fait liée à l’épaisseur de la membrane des divers organites cellulaires [14]. Selon cette hypothèse, la γ-sécrétase cliverait toujours exactement au milieu du domaine transmembranaire mais l’épaisseur plus importante du feuillet membranaire du réticulum endoplasmique entraînerait le « décalage » de l’hydrolyse vers l’extrémité C-terminale, plus en amont au niveau de la liaison 42/43 du peptide Aβ [14]. La voie d’endocytose concernerait la fraction de βAPP ayant échappé aux coupures intracellulaires (voir plus haut). Celle-ci serait internalisée (signalons ici qu’une séquence consensus NPTY de réinternalisation est effectivement localisée au niveau de l’extrémité C-terminale de la βAPP) et subirait les diverses coupures protéolytiques dans le compartiment endosomique précoce. Une partie de la βAPP internalisée semble aussi ciblée vers les compartiments lysosomiques pour sa dégradation terminale (Figure 3).
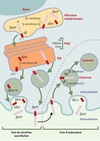 |
Figure 3. Voie amyloïdogénique de maturation de la βAPP. La voie sécrétoire constitutive conduisant à la sécrétion de peptide amyloïde (Aβ) a établi que le peptide Aβ42 était produit très tôt dans le réticulum endoplasmique alors que le peptide Aβ40 apparaissait plus tard, dans le réseau trans-golgien (TGN). Un pool de βAPP échappe à ces coupures intracellulaires et est intégré dans la membrane plasmique. Après endocytose, cette βAPP subit dans les endosomes précoces les coupures engendrant le peptide amyloîde qui est ensuite sécrété. Il apparaît que la βAPP peut aussi être ciblée vers le compartiment lysosomial pour une dégradation finale. |
La nature de la β-sécrétase est maintenant consensuelle. Cinq groupes, presque simultanément, ont décrit deux protéines homologues appelées BACE-1 et -2 (pour β-site APP cleaving enzyme) comme nouvelles protéases acides membranaires présentant la spécificité de type β-sécrétase (pour revue, voir [15]). Il a été montré que la contribution majeure est due à BACE-1 alors que BACE-2, peu présente au niveau cérébral, contribuerait de manière anecdotique à la production de peptide Aβ. Cela a été mis en évidence de manière particulièrement convaincante en invalidant le gène codant pour BACE-1 [16, 17]. Il apparaît que les cellules dérivées de ces souris knock-out ne produisent plus de peptide Aβ. Il faut toutefois souligner que le gène codant pour BACE-2 est porté par le chromosome 21 [18]. Il n’est donc pas impossible que dans les cas de trisomie 21 - toujours caractérisés par les stigmates histopathologiques de la maladie d’Alzheimer dus à l’« extra-copie » du gène de la βAPP (codée elle aussi par le chromosome 21) - la « surproduction » de BACE-2 ne puisse contribuer à l’exacerbation de la production de peptide Aβ.
La nature de la γ-sécrétase est actuellement l’objet d’un débat passionné et passionnant autour des deux protéines, les présénilines 1 et 2, responsables de la majorité des formes familiales à début précoce de maladie d’Alzheimer.
Que sont les présénilines ?
Les présénilines 1 et 2 (PS1/PS2) sont deux protéines, respectivement de 467 et 441 acides aminés, qui présentent 67 % d’homologie (Figure 4) (→).
(→) m/s 1999, n°8/9, p. 1043
 |
Figure 4. Structure et protéolyse des présénilines. La structure présentée, à 8 domaines transmembranaires, est encore discutée. Les sites de coupure par la/les présénilinase(s) et les diverses caspases interviennent au niveau de la grande boucle intracellulaire hydrophile. |
À l’heure actuelle, plus de 70 mutations ponctuelles ont été identifiées sur la PS1 et six sur la PS2, ces nombres continuant actuellement d’augmenter (pour plus de détails, voir le site http://www.alzforum.org/). Les présénilines sont des protéines de membrane possédant un nombre apparemment pair de segments transmembranaines et une large boucle cytoplasmique (Figure 4). Ce sont des protéines présentes dans le système nerveux central, essentiellement dans les neurones. Au niveau subcellulaire, il apparaît que la majorité des présénilines est localisée au niveau du réticulum endoplasmique et du système golgien même si quelques travaux semblent indiquer leur présence au niveau de la membrane plasmique. Les présénilines ne sont ni acétylées ni sulfatées ni glycosylées, mais subissent des processus de phosphorylation qui modulent vraisemblablement leur fonction de contrôle de mort cellulaire (pour revue, voir [19]). Les présénilines sont la cible de diverses activités protéolytiques. Notamment, il a été bien établi qu’elles subissent une coupure protéolytique par une activité que l’on nommera « présénilinase » qui engendre des fragments N-et C-terminaux de respectivement 28-30 et 18-20 kDa. Cette coupure intervient au niveau de la séquence codée par l’exon 10 puisqu’un mutant de délétion de cet exon est totalement résistant à cette protéolyse. Il faut signaler que les holoprotéines de présénilines sont très peu représentées dans le cerveau et/ou, de manière endogène, dans divers systèmes cellulaires, car la coupure par la présénilinase intervient très tôt dans le réticulum endoplasmique. Il apparaît que les produits N- et C-terminaux de protéolyse des présénilines interagissent et forment des hétéromères stables qui correspondent aux complexes biologiquement actifs des présénilines [20]. Plus récemment, il a été établi que les présénilines subissaient aussi de manière alternative une attaque enzymatique par des activités de type caspase [21, 22] qui libérent des fragments C-terminaux capables d’influencer les processus de mort neuronale [23, 24].
Les présénilines présentent des propriétés remarquables de protéines chaperons (pour revue, voir [25]) (→) capables d’interagir avec de nombreuses protéines, et notamment avec la βAPP et avec la protéine Notch (voir plus loin). Ce sont des protéines multifonctionnelles responsables, entre autres, de la modulation des processus précoces de différenciation lors de l’embryogenèse ou, au contraire, d’événements pouvant intervenir beaucoup plus tardivement dans le contrôle de la réponse apoptotique de divers types cellulaires.
→ m/s 2000, n°5, p. 630
Les présénilines contribuent à la maturation physiopathologique de la β APP
Le fait que les présénilines interagissent avec la βAPP est sans doute le premier indice indirect d’un contrôle de la maturation de la βAPP par les présénilines. Cela a été étayé par l’observation que les cerveaux de patients affectés par une maladie d’Alzheimer liée à une mutation portée par la PS1 montraient tous une production exacerbée de peptide Aβ et plus particulièrement d’Aβ42. Cette observation valait aussi pour six cas décrits portant la mutation A141I sur la PS2. Cette augmentation d’Aβ42 a aussi été notée dans le plasma, dans le produit de sécrétion des fibroblastes des sujets affectés par ces mutations, ainsi que dans le cerveau de souris transgéniques surexprimant une PS1 mutée. De manière plus générale, une approche de biologie cellulaire a permis d’établir que toute cellule surexprimant une préséniline mutée présentait une production exacerbée de peptide amyloïde, et que cette augmentation semblait sélectivement affecter la production du peptide « pathogène » de 42 résidus (pour revue, voir [26]). De manière intéressante, nous avons montré que les mutations des présénilines menaient aussi à une altération de la voie physiologique α-sécrétase caractérisée par une baisse de l’APPα sécrété [27].
Il semble que la modulation de la maturation de la βAPP par les mutations pathogènes dérègle un processus normal de contrôle par les présénilines sauvages. En effet, nous avons établi que la surexpression de PS1 et de PS2 conduit à une augmentation de la production d’APPα et d’Aβ40 [27, 28]. Il faut rappeler ici que le peptide Aβ40 est un fragment normal, dérivé de la protéolyse physiologique de la βAPP. Sa surproduction n’est donc ici qu’un indice de la surexpression de la PS1 « normale ». Nous avons établi que les produits C-terminaux des PS1 et des PS2 induisent aussi, comme cela est attendu, le même phénotype que PS1 et PS2 [29].
Les présénilines sauvages et mutées sont rapidement dégradées après ubiquitinylation par le protéasome [28, 30, 31]. De manière intéressante, les inhibiteurs du protéasome potentialisent les deux phénotypes induits par les présénilines normales ou mutées (Figure 5). Quand les cellules expriment les présénilines sauvages, ces inhibiteurs augmentent sélectivement les productions d’APPα et d’Aβ40. Quand les présénilines sont mutées, ces mêmes inhibiteurs potentialisent le phénotype pathogène illustré par la baisse d’APPα et l’augmentation sélective d’Aβ42. Ces résultats nous ont conduits à proposer que les présénilines interagissaient avec la βAPP en amont des sécrétases et que leur fonction était sous le contrôle du protéasome (Figure 5).
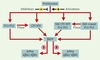 |
Figure 5. Influence des présénilines sur les voies physiopathologiques de maturation de la fiAPP et contrôle par le protéasome. Le protéasome dégrade les présénilines sauvages (PS1/PS2) ou mutées (FAD-linked PS1/PS2) ainsi que les fragments C-terminaux (CTF) et N-terminaux (NTF) dérivés de la maturation des diverses présénilines par la présénilinase (PSase) en amont de leur interaction avec la βAPP, contribuant ainsi à la modulation des produits engendrés par l’α-sécrétase (APPα) et par l’action combinée des β- et des γ-sécrétases (Aβ40 et Aβ42). Les présénilines sauvages contribuent à la production d’APPα et de Aβ40 (voie 1) dont les productions sont potentialisées par les inhibiteurs du protéasome. Les présénilines mutées conduisent à moins d’APPα et à une augmentation du rapport Aβ42/Aβ40 (voie 2). Les inhibiteurs du protéasome exacerbent ce dernier phénotype. Il est spéculé dans ce schéma que l’accélération de la dégradation des présénilines sauvages ou mutées, grâce à des activateurs du protéasome, pourraient conduire à une diminution de la production des peptides Aβ quelle que soit la nature sporadique ou génétique de la maladie d’Alzheimer. |
Les présénilines ne sont pas les γ-sécrétases
Comme cela est évoqué plus haut, la possibilité que les présénilines soient la γ-sécrétase fait l’objet d’un débat majeur dans le domaine de la biologie cellulaire de la maladie d’Alzheimer (pour revues contradictoires, voir [32, 33]). Un des arguments forts en faveur de cette hypothèse consiste notamment en l’observation que l’invalidation des gènes codant pour les présénilines abolit la production de peptide Aβ et la production de NICD (Notch intracellular domain), un produit de la protéolyse d’une autre protéine membranaire appelée Notch [34, 35] (→).
(→) m/s 1999, n°3, p. 414
Ces deux événements catalytiques paraissent bloqués de manière similaire par divers inhibiteurs dont certains, photoréactifs, peuvent apparemment se lier de manière covalente aux présénilines [36, 37]. Enfin, il a été décrit que la mutation de deux résidus aspartyl en position 257 et 385 (séquence de la PS1) conduisait à l’abolition de la production d’Aβ, de NICD et de la coupure endoprotéolytique de type présénilinase des présénilines [38]. Cela a conduit Wolfe et ses collaborateurs à suggérer que les présénilines pourraient être un nouveau type de protéase acide transmembranaire autocatalytiquement activable. Tous ces résultats ont été discutés et parfois totalement contredits dans la littérature. Tout d’abord, il est absolument clair que les événements cataboliques conduisant à la production d’Aβ et de NICD peuvent être pharmacologiquement discriminés. Ainsi, la mutation de l’aspartyl 257 abolit la production de NICD sans affecter la production d’Aβ [39]. Un récent article montre même que les cellules exprimant la PS1 mutée au niveau des deux Asp257/385 produisent autant de peptide Aβ que les présénilines non mutées [40]. Par ailleurs, certaines mutations des présénilines affectent la production de Aβ et de NICD dans des proportions rigoureusement inverses [41]. Nous avons établi que de nouveaux inhibiteurs synthétiques non peptidiques pouvaient bloquer la production de peptide Aβ sans affecter celle de NICD [42]. Nous avons aussi montré récemment que des fibroblastes dans lesquels les gènes de PS1 et PS2 avaient été inactivés produisaient autant de peptides Aβ40 et 42 endogènes sécrétés et intracellulaires que les cellules parentales, alors que la production de NICD était effectivement abolie par l’absence de présénilines [43]. Il faut signaler ici que les études indiquant que la production de peptide Aβ était abolie dans les cellules murines dépourvues de présénilines ont été réalisées après la surexpression de βAPP humaine arborant la mutation suédoise et ne portaient que sur le peptide Aβ total sécrété [34, 35]. Plus troublant encore, un de ces groupes a rapporté que les cellules PSI-/- produisaient des quantités importantes de peptide 2-42 [44]. Il est difficile de concevoir que ces cellules puissent posséder une activité γ-sécrétase spécifiquement impliquée dans la production d’Aβ2-42 mais pas d’Aβ1-42. Puisque la même séquence est ciblée par la γ-sécrétase, il faudrait envisager que la spécificité de coupure soit due à la nature de résidus situés 40 acides aminés en aval ! Il apparaît plus probable que les présénilines modulent la maturation de βAPP via leur activité de type « chaperon ». Il est intéressant, dans cette optique, de noter que Kim et al. ont montré que les mutations des résidus aspartyl des présénilines affectent le ciblage et la stabilité de la βAPP [40].
Le débat concernant la nature de la γ-sécrétase n’est pas uniquement une gymnastique intellectuelle ou une simple querelle de chapelle. Elle conditionne une des stratégies futures visant à ralentir ou à bloquer la production de peptide Aβ. Si la même activité relargue NICD, il apparaît alors probable que cibler la γ-sécrétase via des inhibiteurs spécifiques soit voué à l’échec en regard des effets délétères (notamment sur l’hématopoïèse) attendus d’un blocage de la production de NICD.
Conclusions
Les présénilines font l’objet de toutes les attentions des biologistes qui travaillent sur la maladie d’Alzheimer. Ces protéines multifonctionnelles n’ont pas encore révélé tous leurs secrets mais chaque jour apporte son lot d’éléments nouveaux permettant une meilleure compréhension de leur physiologie. À l’évidence, les présénilines contrôlent la maturation de la βAPP. De manière tout aussi évidente, les mutations de ces protéines accélèrent le processus neurodégénératif lié à la maladie d’Alzheimer. Des stratégies originales comme l’exacerbation de leur dégradation via des activateurs du protéasome, ou des inhibiteurs de l’interaction des présénilines avec la βAPP pourraient être envisagées. Ces pistes volontairement provocatrices et sans doute risquées (le protéasome dégrade de nombreuses protéines et les présénilines interagissent avec des partenaires cellulaires multiples) ne sont sans doute que quelques-unes des options pharmacologiques visant ces protéines et pourraient éventuellement être envisagées comme des approches complémentaires de stratégies thérapeutiques plus classiques visant au blocage des β-et des γ-sécrétases ou à l’activation de l’α-sécrétase.
Références
- Van Broeckhoven C. Presenilins and Alzheimer disease. Nat Genet 1995 ; 11 : 230–2. [Google Scholar]
- Checler F. Presenilins: multifunctional proteins involved in Alzheimer’s disease pathology. IUBMB Life 1999 ; 48 : 33–9. [Google Scholar]
- Checler F. Processing of the β-amyloid precursor protein and its regulation in Alzheimer’s disease. J Neurochem 1995 ; 65 : 1431–44. [Google Scholar]
- Burdick D, Soreghan B, Kwon M, et al. Assembly and aggregation properties of synthetic Alzheimer’s A4/β amyloid peptide analogs. J Biol Chem 1992 ; 267 : 546–54. [Google Scholar]
- Mattson MP. Cellular actions of β-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev 1997 ; 77 : 1081–132. [Google Scholar]
- Octave JN. The amyloid peptide and its precursor in Alzheimer’s disease. Rev Neurosci 1995 ; 6 : 287–316. [Google Scholar]
- Marambaud P, Chevallier N, Ancolio K, Checler F. Post-transcriptional contribution of a cAMP-dependent pathway to the formation of α-and β/γ-secretases-derived products of βAPP maturation in human cells expressing wild type and Swedish mutated βAPP. Mol Med 1998 ; 4 : 715–23. [Google Scholar]
- Vincent B, Paitel E, Saftig P, et al. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbolesters-regulated normal cleavage of the cellular prion protein.J Biol Chem 2001 ; 276 : 37743–6. [Google Scholar]
- Lammich S, Kojro E, Postina R, et al. Constitutive and regulated α-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA 1999 ; 96 : 3922–7. [Google Scholar]
- Lopez-Perez E, Zhang Y, Franck SJ, Creemers J, Seidah N, Checler F. Constitutive α-secretase cleavage of the β-amyloid precursor protein in the furin-deficient LoVo cell line: involvement of the prohormone convertase 7 (PC7) and the disintegrin metalloprotease ADAM10. J Neurochem 2001 ; 76 : 1532–9. [Google Scholar]
- Buxbaum JD, Liu KN, Luo Y, ét al. Evidence that tumor necrosis factor α converting enzyme is involved in regulated α-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem 1998 ; 273 : 27765–7. [Google Scholar]
- Lopez-Perez E, Seidah N, Checler F. A proprotein convertase activity contributes to the processing of the Alzheimer’s β-amyloid precursor protein in human cells: evidence for a rôle of the prohormone convertase PC7 in the constitutive α-secretase pathway. J Neurochem 1999 ; 73 : 2056–62. [Google Scholar]
- Hartmann T, Bieger SC, Bruhl B, et al. Distinct sites of intracellular production for Alzheimer’s disease Aβ40/42 amyloid peptides. Nat Med 1997 ; 3 : 1016–20. [Google Scholar]
- Lichtenthaler SF, Wang R, Grimm H, Uljon SN, Masters CL, Beyreuther K. Mechanism of the cleavage specificity of Alzheimer’s disease γ-secretase identified by phenylalanine-scanning mutagenesis of the transmembrane domain of the amyloid precursor protein. Proc Natl Acad Sci USA 1999 ; 96 : 3053–8. [Google Scholar]
- Vassar R, Citron M. Aβ-generating enzymes: recent advances in γ- and γ-secretases research. Neuron 2000 ; 27 : 419–22. [Google Scholar]
- Cai H, Wang Y, Mc Carthy D, et al. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat Neurosci 2001 ; 4 : 233–4. [Google Scholar]
- Luo Y, Bolon B, Kahn S, et al Mice deficient in BACE1, the Alzheimer’s β-secretase, have normal phenotype and abolished β-amyloid generation. Nat Neurosci 2001 ; 4 : 231–2. [Google Scholar]
- Saunders AJ, Kim TW, Tanzi RE. BACE maps to chromosome 11 and a BACE homolog, BACE2, reside in the obligate down syndrome region of chromosome 21. Science 1999 ; 286 : 1254–5. [Google Scholar]
- Checler F. Presenilins: structural aspects and post-translational events. Mol Neurobiol 1999 ; 19 : 255–65. [Google Scholar]
- Capell A, Grünberg J, Pesold B, et al. The proteolytic fragments of the Alzheimer’s disease-associated presenilin-1 form heterodimers and occur as a 100-150-kDa molecular mass complex. J Biol Chem 1998 ; 273 : 3205–11. [Google Scholar]
- Loetscher H, Deuschle U, Brockhaus M, et al. Presenilins are processed by caspase-like proteases. J Biol Chem 1997 ; 272 : 20655–9. [Google Scholar]
- Kim TW, Pettingell WH, Jung YK, Kovacs DM, Tanzi RE. Alternative cleavage of Alzheimer-associated presenilins during apoptosis by a caspase-3 family protease. Science 1997 ; 277 : 373–6. [Google Scholar]
- Vito P, Lacana E, D’Adamio L. Interfering with apoptosis: Ca2+-binding protein ALG-2 and Alzheimer’s disease gene ALG-3. Science 1996 ; 271 : 521–5. [Google Scholar]
- Grünberg J, Walter J, Loetscher H, Deuschle U, Jacobsen H, Haass C. Alzheimer’s disease associated presenilin-1 holoprotein and its 18-20 kDa C-terminal fragment are death substrates for proteases of the caspase family. Biochemistry 1998 ; 37 : 2263–70. [Google Scholar]
- Van Gassen G, Annaert W, Van Broekhoven C. Binding partners of Alzheimer’s disease proteins: are they physiologically relevant? Neurobiol Dis 2000 ; 7 : 135–51. [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins and therapy. Physiol Rev 2001 ; 81 : 741–66. [Google Scholar]
- Ancolio K, Marambaud P, Dauch P, Checler F. α-secretase-derived product of β-amyloid precursor protein is decreased by presenilin 1 mutations linked to familial Alzheimer’s disease. J Neurochem 1997 ; 69 : 2494–9. [Google Scholar]
- Marambaud P, Chevallier N, Barelli H, Wilk S, Checler F. Proteasome contributes to the α-secretase pathway of amyloid precursor protein in human cells. J Neurochem 1997; 68 : 698–703. [Google Scholar]
- Alves da Costa C, Ancolio K, Checler F. C-terminal maturation fragments of presenilin 1 and 2 control secretion of APPα and Aβ by human cells and are degraded by the proteasome. Mol Med 1999 ; 5 : 160–8. [Google Scholar]
- Marambaud P, Alves da Costa C, Ancolio K, Checler F. Alzheimer’s disease-linked mutation of presenilin 2 (N141I-PS2) drastically lowers APPα secretion: control by the proteasome. Biochem Biophys Res Commun 1998 ; 252 : 134–8. [Google Scholar]
- Kim TW, Pettingell WH, Hallmark OG, Moir RD, Wasco W, Tanzi, RE. Endoproteolytic cleavage and proteasomal degradation of presenilin 2 in transfected cells. J Biol Chem 1997 ; 272 : 11006–10. [Google Scholar]
- Checler F. The multiple paradoxes of presenilins. J Neurochem 2001 ; 76 : 1621–7. [Google Scholar]
- Wolfe MS. Presenilins and γ-secretase: structure meets function. J Neurochem 2001 ; 76 : 1615–20. [Google Scholar]
- Herreman A, Serneels L, Annaert W, Collen D, Schoonjans L, De Strooper B. Total inactivation of γ-secretase activity in presenilin-deficient embryonic stem cells. Nat Cell Biol 2000 ; 2 : 461–2. [Google Scholar]
- Zhang Z, Nadeau P, Song W, et al. Presenilins are required for γ-secretase cleavage of βAPP and transmembrane cleavage of Notch-1. Nat Cell Biol 2000 ; 2 : 463–5. [Google Scholar]
- Li YM, Xu M, Lai MT, et al. Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 2000 ; 405 : 689–94. [Google Scholar]
- Esler WP, Kimberly WT, Ostaszewski BL, et al. Transition-state analogue inhibitors of γ-secretase bind directly to presenilin-1. Nat Cell Biol 2000 ; 2 : 428–34. [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe, DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 1999 ; 398 : 513–7. [Google Scholar]
- Capell A, Steiner H, Romig H, et al. Presenilin-1 differentially facilitates endoproteolysis of the β-amyloid precursor protein and Notch. Nat Cell Biol 2000 ; 2 : 205–11. [Google Scholar]
- Kim SH, Leem JY, Lah JJ, et al. Multiple effects of aspartate mutant presenilin 1 on the processing and trafficking of amyloid precursor protein. J Biol Chem 2001 ; 276 : 43343–50. [Google Scholar]
- Kulic L, Walter J, Multhaup G, et al. Separation of presenilin function in amyloid β-peptide generation and endoproteolysis of Notch. Proc Natl Acad Sci USA 2000 ; 97 : 5913–8. [Google Scholar]
- Petit A, Bihel F, Alves da Costa C, Pourquié O, Kraus JL, Checler F. New protease inhibitors prevent γ-secretase-mediated Aβ40/42 production without affecting Notch cleavage. Nat Cell Biol 2001 ; 3 : 507–11. [Google Scholar]
- Armogida M, Petit A. Vincent B, Scarzello S, Alves da Costa C, Checler F. Endogenous β-amyloid production in presenilin-deficient embryonic mouse fibroblasts. Nat Cell Biol 2001 ; 3 : 1030–3. [Google Scholar]
- Wiltfang J, Esselmann H, Cupers P, et al. Elevation of β-amyloid peptide 2-42 in sporadic and familial Alzheimer’s disease and its generation in PS1 knockout cells. J Biol Chem 2001 ; 276 : 42645–57. [Google Scholar]
Liste des figures
|
Figure 1. Structure schématique de la βAPP et mutations pathogènes. Structure transmembranaire de la βAPP ciblée par les β-et γ-sécrétases et engendrant le peptide Aβ. Une coupure alternative par l’α-sécrétase intervient dans la séquence Aβ et libère un fragment sécrété, l’APPα. Les principales mutations intervenant au niveau de la séquence Aβ sont représentées. |
| Dans le texte | |
|
Figure 2. Voie non amyloïdogénique de maturation de la βAPP. L a βAPP est coupée dans le réseau trans-golgien (TGN) par une α-sécrétase intracellulaire. Cette coupure libère un fragment, l’APPα, qui est engendré par la voie sécrétoire constitutive ou réglé par la protéine kinase C. Un pool de βAPP échappe à cette coupure intracellulaire et est intégré dans la membrane plasmique où une α-sécrétase membranaire engendre la même coupure. Cette voie de maturation de la βAPP est considérée comme non amyloïdogénique puisque les α-sécrétases interviennent au milieu du peptide amyloïde (Aβ, segment représenté en rouge), prévenant ainsi sa formation. |
| Dans le texte | |
|
Figure 3. Voie amyloïdogénique de maturation de la βAPP. La voie sécrétoire constitutive conduisant à la sécrétion de peptide amyloïde (Aβ) a établi que le peptide Aβ42 était produit très tôt dans le réticulum endoplasmique alors que le peptide Aβ40 apparaissait plus tard, dans le réseau trans-golgien (TGN). Un pool de βAPP échappe à ces coupures intracellulaires et est intégré dans la membrane plasmique. Après endocytose, cette βAPP subit dans les endosomes précoces les coupures engendrant le peptide amyloîde qui est ensuite sécrété. Il apparaît que la βAPP peut aussi être ciblée vers le compartiment lysosomial pour une dégradation finale. |
| Dans le texte | |
|
Figure 4. Structure et protéolyse des présénilines. La structure présentée, à 8 domaines transmembranaires, est encore discutée. Les sites de coupure par la/les présénilinase(s) et les diverses caspases interviennent au niveau de la grande boucle intracellulaire hydrophile. |
| Dans le texte | |
|
Figure 5. Influence des présénilines sur les voies physiopathologiques de maturation de la fiAPP et contrôle par le protéasome. Le protéasome dégrade les présénilines sauvages (PS1/PS2) ou mutées (FAD-linked PS1/PS2) ainsi que les fragments C-terminaux (CTF) et N-terminaux (NTF) dérivés de la maturation des diverses présénilines par la présénilinase (PSase) en amont de leur interaction avec la βAPP, contribuant ainsi à la modulation des produits engendrés par l’α-sécrétase (APPα) et par l’action combinée des β- et des γ-sécrétases (Aβ40 et Aβ42). Les présénilines sauvages contribuent à la production d’APPα et de Aβ40 (voie 1) dont les productions sont potentialisées par les inhibiteurs du protéasome. Les présénilines mutées conduisent à moins d’APPα et à une augmentation du rapport Aβ42/Aβ40 (voie 2). Les inhibiteurs du protéasome exacerbent ce dernier phénotype. Il est spéculé dans ce schéma que l’accélération de la dégradation des présénilines sauvages ou mutées, grâce à des activateurs du protéasome, pourraient conduire à une diminution de la production des peptides Aβ quelle que soit la nature sporadique ou génétique de la maladie d’Alzheimer. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.