")
")
| Issue |
Med Sci (Paris)
Volume 17, Number 11, Novembre 2001
|
|
|---|---|---|
| Page(s) | 1112 - 1119 | |
| Section | Articles de Synthèse | |
| DOI | https://doi.org/10.1051/medsci/200117111112 | |
| Published online | 15 novembre 2001 | |
IL-12 et IFN-γ : un axe clé de l’immunité anti-mycobactérienne chez l’homme
IL-12 and IFN-γ: two key factors of anti-mycobacterial immunity in humans
Laboratoire de génétique humaine des maladies infectieuses, Unité mixte de recherche, Université de Paris René-Descartes-Inserm U. 550, Faculté de médecine Necker, 156, rue de Vaugirard, 75015 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les maladies mycobactériennes communes, la tuberculose, la lèpre, et l’ulcère de Buruli, sont responsables d’une morbidité et d’une mortalité considérables dans le monde. Les autres mycobactéries, comme les souches vaccinales de Calmette et Guérin (BCG) et les mycobactéries environnementales, sont moins virulentes chez l’homme mais peuvent être responsables d’infections sévères si la réponse immunitaire est imparfaite. Chez l’homme, l’étude du syndrome de prédisposition mendélienne aux infections mycobactériennes, a permis d’établir le rôle clé de l’interféron-γ (IFN-γ), mais également de son principal inducteur l’interleukine-12 (IL-12), dans les mécanismes moléculaires de l’immunité anti-mycobactérienne. Le rôle de cet axe dans la vulnérabilité aux infections mycobactériennes communes peut à présent être testé.
Abstract
Common mycobacterial diseases, tuberculosis, leprosy, and Buruli ulcer are responsible for considerable mortality and morbidity world-wide. Other mycobacterial species, such as Bacillus Calmette-Guerin (BCG) vaccines and environmental mycobacteria, are less virulent in humans, although they may cause severe infections in case of impaired immunity. The clinical characterisation and molecular elucidation of the syndrome of Mendelian susceptibility to mycobacterial infections demonstrated the key role of Interferon-γ (IFN-γ), and its potent inducer Interleukin-12 (IL-12), in anti-mycobacterial immunity. The role of the IFN-γ/IL-12 axis in vulnerability to common mycobacterial diseases is now testable.
© 2001 médecine/sciences - Inserm / SRMS
Le genre Mycobacterium, comprend des bactéries aérobies qui ont une paroi très particulière et riche en lipides [1]. De nombreuses espèces de mycobactéries peuvent être distinguées, certaines responsables de maladies communes comme M. leprae, responsable de la lèpre, mais aussi le complexe Mycobacterium tuberculosis comprenant M. tuberculosis, M. bovis, et M. africanum responsables de la tuberculose chez l’homme, ou encore M. ulcerans responsable de l’ulcère de Buruli [2]. D’autres sont moins virulentes, telles que les bacilles de Calmette et Guérin (BCG), souches atténuées de M. bovis [3], et couramment utilisées pour la vaccination contre la tuberculose et la lèpre, ou bien encore les mycobactéries dites environnementales, ou non tuberculeuses (MNT), et retrouvées de façon ubiquitaire [4].
De nombreuses études chez la souris ont permis de caractériser le rôle indispensable des lymphocytes T, des macrophages et de différentes cytokines dans le développement d’une immunité protectrice contre les infections mycobactériennes [5]. En particulier, l’interaction de lymphocytes T CD4, et dans une moindre mesure CD8, avec des macrophages infectés, apparaît déterminant pour l’élimination de la mycobactérie [6]. Dans le cadre de la réponse anti-mycobactérienne, cette interaction est facilitée par la mise en place de granulomes au-niveau du site infectieux. Il s’agit de structures multicellulaires composées de lymphocytes, de macrophages infectés, de cellules épithélioïdes et de cellules géantes de Langhans issues respectivement de la différenciation et de la fusion de macrophages. Ces granulomes se forment en particulier grâce au recrutement de lymphocytes au cours de la réaction inflammatoire consécutive à l’infection [7]. Parmi les cytokines participant à l’interaction lymphocyte/macrophage, le rôle déterminant de l’IL-12, mais également de l’IFN-γ et du TNF-α, a été clairement démontré chez la souris. En effet, les souris dont les gènes de ces trois cytokines ou de leurs récepteurs ont été invalidés sont très vulnérables aux infections mycobactériennes, et ne développent pas de granulomes [5].
Chez l’homme, le rôle des lymphocytes T dans l’immunité anti-mycobactérienne a également été constaté. Les individus présentant un déficit lymphocytaire T majeur, acquis [8], ou héréditaire [9], sont en effet très sensibles aux infections mycobactériennes. Une sensibilité comparable a également été constatée chez les individus présentant un déficit des phagocytes [9]. En revanche, à la différence du système murin, une coopération cellulaire impliquant les lymphocytes T, et aboutissant à l’activation des mécanismes de destruction intracellulaire des mycobactéries par les macrophages infectés n’a jamais été clairement démontrée chez l’homme [5]. L’étude récente de patients présentant un syndrome de prédisposition mendélienne aux infections mycobactériennes (MIM 209950), a permis de mieux caractériser les mécanismes moléculaires de l’immunité anti-mycobactérienne chez l’homme. Ces patients ont une vulnérabilité héréditaire aux infections mycobactériennes, et dans une moindre mesure aux salmonelles, en l’absence de tout déficit immunitaire connu [10, 11]. La caractérisation moléculaire de ces patients a permis d’identifier des déficits dans cinq gènes participant aux voies d’activation cellulaire par l’IFN-γ ou l’IL-12 (figure 1), mettant ainsi en évidence le rôle indispensable de l’IFN-γ et de son principal inducteur, l’IL-12 dans le contrôle des infections mycobactériennes chez l’homme.
 |
Figure 1. Étiologie génétique du syndrome de prédisposition mendélienne aux infections mycobactériennes. |

Rôle de l’immunité induite par l’IFN-γ
L’IFN-γ est une cytokine homodimérique produite principalement par les lymphocytes T et NK. Son récepteur, qui est exprimé par la majorité des types cellulaires, se compose d’une chaîne α (ou IFN-γR1), qui permet la fixation de l’IFN-γ, et d’une chaîne β (ou IFN-γR2) ne jouant pas directement de rôle dans la fixation du ligand, mais qui permet la transduction du signal [12]. Les deux chaînes IFN-γR1 et IFN-γR2 sont constitutivement associées à deux kinases, JAK1 et JAK2 respectivement, au niveau de leur domaine intracellulaire (figure 2). Lors de la fixation de l’IFN-γ, il y a rapprochement des deux chaînes du récepteur entraînant la phosphorylation réciproque de JAK1 et JAK2, ainsi que la phosphorylation du site de fixation de la molécule STAT1 sur la chaîne IFN-γR1. Cette fixation entraîne la phosphorylation de la molécule STAT1 qui s’homodimérise, formant ainsi le complexe GAF, et traverse la paroi nucléaire pour aller se fixer sur les sites GAS (IFN-γ activation sequence) au niveau des promoteurs des gènes inductibles par l’IFN-γ [12] (figure 2).
 |
Figure 2. Voie de signalisation de l’IFN-γ. |
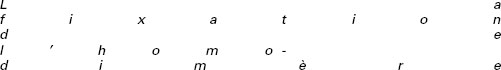
Différentes mutations responsables d’un défaut complet de la chaîne IFN-γR1 [13-19], et une mutation entraînant un défaut complet de la chaîne IFN-γR2 [20], ont été identifiées chez des individus présentant le syndrome de prédisposition mendélienne aux infections mycobactériennes. A l’échelle cellulaire, ces différentes mutations sont responsables d’une perte totale de réponse à l’IFN-γ. Sur le plan clinique, tous les patients atteints d’un défaut complet des molécules IFN-γR1 ou IFN-γR2, ont développé des infections sèvères par le BCG ou les mycobactéries environnementales, et présenté des structures granulomateuses mal circonscrites et peu différenciées. Comme chez la souris, l’IFN-γ paraît donc avoir un rôle indispensable dans l’immunité anti-mycobactérienne et dans la formation de granulomes chez l’homme. De plus, les patients présentant un défaut complet des molécules IFN-γR1 ou IFN-γR2 ont des taux élevés d’IFN-γ dans le sérum. Ce résultat est probablement dû à une accumulation de l’IFN-γ produit en réponse à l’emballement de l’infection mycobactérienne, et qui ne peut pas être capté par les cellules de ces patients qui ont un récepteur non fonctionnel [21]. Cette caractéristique apparaît également très intéressante d’un point de vue clinique, puisqu’elle permet un diagnostic rapide des défauts complets en récepteur de l’IFN-γ, par simple dosage sérique de la cytokine.
Des défauts partiels, récessifs [22, 23] ou dominants [24, 25] de l’IFN-γR1, mais également un défaut partiel récessif de l’IFN-γR2 [26], entraînant une diminution de la réponse cellulaire à l’IFN-γ, ont été identifiés chez d’autres individus présentant le syndrome. Ces patients ont un phénotype clinique atténué, caractérisé par des infections curables et la présence de granulomes bien différenciés. Ces résultats indiquent qu’il existe une forte corrélation entre le génotype et le phénotype cellulaire et clinique chez les individus atteints de ce syndrome. Les infections mycobactériennes sont sévères chez les individus présentant des défauts complets d’activation par l’IFN-γ, et plus modestes chez les patients présentant des défauts partiels d’activation par l’IFN-γ. Il apparaît, de plus, une grande hétérogénéité allélique dans les défauts en IFN-γR1, avec trois formes récessives (défaut complet avec expression, défaut complet sans expression, défaut partiel) et une forme dominante (défaut partiel avec accumulation d’une molécule IFN-γR1 non fonctionnelle) (figure 3). Plus récemment un défaut partiel de la molécule STAT1 a été identifié chez deux patients présentant une forme atténuée du syndrome, et aucun défaut des molécules IFN-γR1 ou IFN-γR2. Du point de vue clinique et physiopathologique, ils ressemblent aux patients présentant un défaut partiel d’une des deux sousunités du récepteur de l’IFN-γ, avec une infection finalement maîtrisée, et une réponse partielle à l’IFN-γ [27]. Cette mutation bloque la phosphorylation de la protéine STAT1 en réponse à l’IFN-γ, ce qui entraîne un défaut de translocation nucléaire de l’homodimère activateur STAT1/STAT1 (GAF) [27]. Seul le quart des dimères STAT1 qui peuvent se former sont fonctionnels chez ces deux patients, entraînant ainsi un défaut partiel de réponse à l’IFN-γ. Au plan immunologique, ce déficit immunitaire affectant un des médiateurs de l’immunité induite par l’IFN-γ, souligne encore une fois le rôle indispensable de l’IFN-γ dans la réponse anti-mycobactérienne. Il démontre de plus que le contrôle des infections mycobactériennes est non seulement dépendante de l’IFN-γ, mais également que les mécanismes induits par l’IFN-γ passent par l’activation du facteur de transcription STAT1 dans le cadre des dimères GAF.
 |
Figure 3. Hétérogénéité allélique des défauts en IFN-γR1. |

L’immunité induite par l’IFN-γ est donc un trait quantitatif qui détermine l’issue d’une invasion mycobactérienne chez l’homme [28]. Cela suggère qu’il n’existe pas de voie alternative à l’activation cellulaire par l’IFN-γ. Bien qu’il soit difficile de déterminer la fonctionalité des granulomes chez les patients présentant un défaut partiel, il semble cependant incontestable que la formation d’un granulome bien différencié soit insuffisante au contrôle total de l’infection mycobactérienne. Plusieurs questions restent cependant en suspens. Le récepteur de l’IFN-γ étant ubiquitaire, il est difficile de savoir quelles sont la ou les cellules responsables du phénotype clinique chez ces patients. De même, il est difficile à ce stade de déterminer la (ou les) fonction(s) induite(s) par l’IFN-γ, et dont le défaut est responsable de l’absence de granulomes bien différenciés. Contrairement à la souris, l’immunité induite par l’IFN-γ semble, chez l’homme, uniquement consacrée au contrôle des infections mycobactériennes, aucune sensibilité particulière à d’autres pathogènes n’ayant jamais été décrite chez ces patients [29].
Rôle de l’immunité induite par l’IL-12
L’IFN-γ, et les molécules impliquées dans la réponse cellulaire à cette cytokine étant indispensables à l’immunité anti-mycobactérienne, un rôle des molécules activatrices de la production d’IFN-γ, et en particulier de l’IL-12, a donc été recherché chez les patients présentant le syndrome et aucune altération de l’immunité induite par l’IFN-γ. L’IL-12 est une cytokine hétérodimérique composée de deux chaînes (p35 et p40). Principalement sécrétée par les cellules dendritiques et les phagocytes, sa principale fonction apparaît être l’induction de la production d’IFN-γ [30]. Son récepteur, présent sur les lymphocytes T et NK activés (figure 4), se compose de deux chaînes (IL-12Rβ1 et IL-12Rβ2). La co-expression de ces deux chaînes est nécessaire à la fixation de haute affinité de l’IL-12 [31]. Deux kinases appartenant à la famille des Janus kinases (JAK) sont associées aux chaînes IL-12Rβ1 et IL-12Rβ2, il s’agit respectivement de Tyk2 et de Jak2 [32] (figure 4). La fixation de l’IL-12 à son récepteur entraîne la phosphorylation de ces kinases, qui phosphorylent à leur tour le site de liaison de la molécule STAT4 sur le récepteur [33]. La fixation de l’IL-12 à son récepteur entraîne l’activation de trois molécules STAT : STAT1, STAT3 et STAT4, mais seule STAT4 est activée directement via le récepteur de l’IL-12 [34]. La molécule STAT4 activée est dimérisée et transloquée dans le noyau où elle active la transcription du gène IFNG [35].
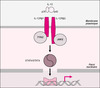 |
Figure 4. Voie de signalisation de l’IL-12. |
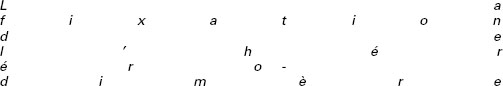
Des patients présentant des défauts complets en IL-12 p40 [36] ou en IL-12Rβ1 [37-41], ont été identifiés. Comme chez la souris, le phénotype clinique et histologique de ces patients est plus atténué que celui des patients présentant un défaut complet de la voie d’activation par l’IFN-γ, avec des infections traitées par antibiothérapie seule, ou antibiothérapie et IFN-γ. Ces patients sont ainsi plutôt comparables aux individus présentant un défaut partiel d’activation par l’IFN-γ, avec la formation de granulomes bien circonscrits et bien différentiés [38, 39]. Bien que des granulomes bien différenciés soient présents, il semble que leur cinétique d’apparition soit retardée en absence d’activation par l’IL-12. En effet, des biopsies effectuées peu après l’infection montraient des régions dépourvues de granulomes différenciés chez la patiente présentant un défaut complet en IL-12 p40, alors que de nombreux granulomes bien différenciés étaient visibles sur une biopsie effectuée plus tardivement sur la même patiente [36]. De façon comparable aux patients présentant un défaut d’activation cellulaire par l’IFN-γ, et à la différence des souris déficientes dans cette même voie d’activation, ces patients ont une sensibilité qui se limite aux infections mycobactériennes, à l’exception de quelques infections par Salmonella.
Au niveau immunologique, le principal défaut constaté chez ces patients est une forte diminution de la production d’IFN-γ pouvant être complémentée in vitro, et de façon dépendante de la dose par ajout d’IL-12 recombinante, confirmant le lien de cause à effet entre le défaut génétique d’IL-12, et la diminution de production d’IFN-γ. La production résiduelle d’IFN-γ par des lymphocytes T et NK de ces patients activés in vitro, rend vraisemblablement compte de leur phénotype clinique atténué, comme le montre également l’effet bénéfique du traitement par l’IFN-γ des patients [42]. Une incertitude demeure cependant sur le type cellulaire responsable de cette production résiduelle d’IFN-γ. Seule l’identification de défauts complets du récepteur de l’IL-12 affectant spécifiquement soit les lymphocytes T, soit les NK, permettrait de discriminer leur participation relative à cette production d’IFN-γ.
Une famille dont un enfant présentait un défaut complet en IL-12Rβ1, et a développé une infection disséminée par le BCG consécutive à sa vaccination, a récemment été étudiée. De façon inattendue, une de ses sœeurs qui présentait la même mutation homozygote du gène IL12Rβ1, a résisté à trois vaccinations par le BCG à différents âges [41]. L’étude de cette patiente semble indiquer, qu’à la différence des individus présentant un défaut d’activation cellulaire par l’IFN-γ, il n’y a pas de corrélation phénotype/génotype dans les défauts d’activation par l’IL-12. En effet, cette observation démontre que des mécanismes indépendants de l’IL-12 peuvent, dans certains cas, suffire au contrôle des infections par des mycobactéries peu virulentes comme le BCG. De façon intéressante, cette patiente a développé à l’adolescence une péritonite tuberculeuse, une forme rare et particulièrement sévère d’infection par M. tuberculosis [43]. L’immunité indépendante de l’IL-12 apparaît ainsi insuffisante pour le contrôle de l’infection par des mycobactéries plus virulentes comme M. tuberculosis [41].
Les mécanismes rendant compte de cette hétérogénéité inter- et intrafamiliale des déficits en IL-12Rβ1 ne sont pas clairs. Une variabilité des facteurs environnementaux et une variabilité des souches vaccinales utilisées chez ces deux enfants ne peuvent pas être exclues, mais sont difficilement analysables a posteriori. Le rôle de variations inter-individuelles de la production d’IFN-γ résiduelle qui serait, comme cela a été proposée par certains auteurs, induite par l’IL-12, mais indépendante de l’IL-12Rβ1 [44], semble peu probable, puisque le phénotype clinique n’est pas moins sévère que chez les patients présentant un défaut en IL- 12 p40. Il paraît plus vraisemblable que ces variations soient dues à une hétérogénéité de la production d’IFN-γ, indépendante de l’IL-12, ou bien à une hétérogénéité de réponse cellulaire à l’IFN-γ, bien que ces hypothèses restent à démontrer.
A la différence de la souris, chez laquelle le rôle indispensable de l’axe IFN-γ/IL-12 dans le contrôle de l’infection par M. tuberculosis avait été clairement démontré, seuls des arguments indirects plaidaient en faveur d’un rôle de ces deux cytokines dans l’immunité contre la tuberculose chez l’homme. En particulier, le rôle de l’immunité induite par l’IL-12 dans la réponse à la tuberculose chez l’homme avait seulement été suggéré dans des études sur la production d’IL-12 [45] et l’expression de son récepteur [46] au niveau du site de l’infection par M. tuberculosis. Cependant, aucun lien de cause à effet n’avait pu être établi entre l’expression de cette cytokine et de son récepteur au niveau du site infectieux, et l’immunité anti-tuberculeuse. Grâce à l’étude de patients présentant le syndrome de prédisposition mendélienne aux infections mycobactériennes, un rôle de l’IFN-γ avait déjà été évoqué par l’étude d’un premier patient présentant un défaut partiel de l’IFN-γR1, qui avait développé les symptômes d’une tuberculose clinique primaire [22]. Ce malade n’avait pas été vacciné par le BCG, ce qui le rend difficilement comparable à la patiente ayant développé une péritonite tuberculeuse [41]. La caractérisation de ces deux patients apporte la première démonstration de l’importance de l’immunité induite par l’axe IL-12/IFN-γ dans le contrôle de l’infection par M. tuberculosis chez l’homme [41].
Conclusions
L’identification et la caractérisation de patients présentant différentes mutations dans les gènes IFNGR1, IFNGR2, STAT1, IL12B, et IL12RB1 a permis de démontrer le rôle essentiel de l’immunité induite par l’IFN-γ, dépendante de l’IL-12, dans le contrôle des infections mycobactériennes chez l’homme. L’efficacité de l’activation cellulaire induite par l’IFN-γ apparaît directement proportionnelle à la sévérité du phénotype clinique observé (Tableau I). Un défaut complet d’activation par l’IL-12, qui se traduit par une production résiduelle d’IFN-γ, permettant donc une activation partielle de l’immunité induite par l’IFN-γ, est lié dans la majorité des cas identifiés à ce jour, au développement d’infections atténuées par les mycobactéries peu virulentes. Cependant, des variabilités phénotypiques inter- et intra-individuelles ont pu être démontrées par l’étude de patients présentant un même génotype IL12RB1. Il semblerait donc que certains mécanismes de l’immunité anti-mycobactérienne indépendants du récepteur de l’IL-12, permettent dans certains cas le contrôle de l’infection par les mycobactéries peu virulentes chez l’homme (Tableau I). Ces mécanismes alternatifs, qui restent cependant à élucider, demeurent insuffisants au contrôle de l’infection par des mycobactéries plus virulentes comme M. tuberculosis. Le rôle essentiel de l’axe IL-12/IFN-γ chez l’homme ressort clairement de l’étude du syndrome de prédispositon mendélienne aux infections mycobactériennes. Une question importante est de savoir qui de l’IFN-γ ou de l’IL-12 apparaît le premier en réponse à l’infection, et induit la production de la seconde cytokine. La production d’IL-12 par les macrophages peu après l’infection est généralement considérée comme l’événement premier de la réponse aux pathogènes à développement intracellulaire. Il a cependant été montré que la production d’IL-12 in vivo était obtenue en réponse à l’infection par le BCG chez des souris immunocompétentes, mais pas chez des souris dépourvues de récepteurs de l’IFN-γ [47]. Des résultats comparables ont été obtenus chez des patients présentant un défaut du récepteur de l’IFN-γ, qui ne produisent que 10 % de la quantité normale d’IL-12 en réponse à l’activation par la phytohémaglutinine (PHA) in vitro [14]. Une production normale d’IL- 12 peut cependant être obtenue après infection de souris déficientes en IFN-γ, par Listeria monocytogenes [48]. Bien qu’il paraisse ainsi difficile de généraliser l’ordre de production de ces deux cytokines dans le cadre d’infections par tous les pathogènes à développement intracellulaire, il semble en tout cas que dans le cadre des infections mycobactériennes, l’IFN-γ soit la première cytokine produite. L’IL-12 et l’IFN-γ doivent donc être considérées comme un axe non pas uni-, mais bidirectionnel, tout au moins dans l’immunité anti-mycobactérienne.
Quatre groupes d’individus peuvent être distingués selon la corrélation entre le génotype et le phénotype clinique et cellulaire.
En tout état de cause, il est tentant de spéculer quant au rôle de mutations entraînant un défaut partiel d’activation cellulaire par l’IL-12, qui n’affecteraient pas l’immunité contre les mycobactéries peu virulentes, mais favoriseraient le développement d’infections par des mycobactéries virulentes comme M. tuberculosis. Il a été estimé qu’environ un tiers de la population mondiale est infecté par M. tuberculosis. Un récent rapport de l’Organisation Mondiale de la Santé (OMS) estime à environ 8 millions le nombre d’individus développant une maladie clinique chaque année, et à 1,9 millions le nombre de décès liés à cette infection [49]. Quelles sont les différences génétiques entre les individus succombant à la maladie, ceux qui développent une maladie curable, et ceux qui sont infectés mais asymptomatiques [50] ? Il est possible qu’il existe un lien direct entre un génotype particulier, impliqué dans la réponse immunitaire contrôlée par l’axe IL-12/IFN-γ, et les différents phénotypes cliniques observés dans le cadre de la tuberculose. L’identification non seulement de nouvelles mutations rares pathogènes de ces gènes, mais également de mutations plus fréquentes ayant un caractère pathogène moins marqué, et leur association aux formes cliniques des infections mycobactériennes communes devrait permettre de tester expérimentalement cette hypothèse .
Références
- Salfinger M. Characteristics of the various species of Mycobacteria. In : Rom WN, Garay SM, eds. Tuberculosis. New York : Little Brown and Company Inc, 1996 : 161–70. [Google Scholar]
- Wolinsky E. Mycobacteria. In : Davis BD, Dulbecco R, Eisen HN, Ginsberg HS, eds. Microbiology, 4th ed. Philadelphia : J.B. Lippincott Company, 1990 : 647–64. [Google Scholar]
- Calmette A.La vaccination préventive contre la tuberculose par le BCG. Paris : Masson, 1927. [Google Scholar]
- Holland SM. Nontuberculous mycobacteria. Am J Med Sci 2001; 321 : 49–55. [Google Scholar]
- Flynn JL, Chan J. Immunology of tuberculosis. Annu Rev Immunol 2001; 19 : 93–129. [Google Scholar]
- Stenger S, Modlin RL. T cell mediated immunity to Mycobacterium tuberculosis. Curr Opin Microbiol 1999; 2 : 89–93. [Google Scholar]
- Kobayashi K, Yoshida T. The immunopathogenesis of granulomatous inflammation induced by Mycobacterium tuberculosis. Meth Enzymol 1996; 9 : 204–14. [Google Scholar]
- Drobniewski F, Pozniak A, Uttley A. Tuberculosis and AIDS. J Med Microbiol 1995; 43 : 85–91. [Google Scholar]
- Primary immunodeficiency diseases. Report of an IUIS Scientific Committee. International Union of Immunological Societies. Clin Exp Immunol 1999; 118 (suppl 1) : 1–28. [Google Scholar]
- Casanova JL, Jouanguy E, Lamhamedi S, Blanche S, Fischer A. Immunological conditions of children with BCG disseminated infection.Lancet 1995; 346 : 581. [Google Scholar]
- Casanova L, Blanche S, Emile JF, et al. Idiopathic disseminated bacillus Calmette-Guerin infection : a French national retrospective study. Pediatrics 1996; 98 : 774–8. [Google Scholar]
- Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor : a paradigm for cytokine receptor signaling. Annu Rev Immunol 1997; 15 : 563–91. [Google Scholar]
- Altare F, Jouanguy E, Lamhamedi-Cherradi S, et al. A causative relationship between mutant IFNγR1 alleles and impaired cellular response to IFN gamma in a compound heterozygous child. Am J Hum Genet 1998; 62 : 723–6. [Google Scholar]
- Holland SA, Dorman SE, Kwon A, et al . Abnormal regulation of interferon gamma, interleukin 12, and tumor necrosis factor alpha in interferon gamma receptor 1 deficiency. J Infect Dis 1998; 178 : 1095–104. [Google Scholar]
- Jouanguy E, Altare F, Lamhamedi S,et al .Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guerin infection. N Engl J Med 1996; 335 : 1956–61. [Google Scholar]
- Newport MJ, Huxley CM, Huston S, et al. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med 1996; 335 : 1941–9. [Google Scholar]
- Pierre-Audigier C, Jouanguy E, Lamhamedi S, et al. Fatal disseminated Mycobacterium smegmatis infection in a child with inherited interferon gamma receptor deficiency. Clin Infect Dis 1997; 24 : 982–4. [Google Scholar]
- Roesler J, Kofink B, Wendisch J, et al. Recurrent mycobacterial and Listeria infections in a child with interferon γ receptor deficiency. Exp Hematol 1999; 27 : 1368–74. [Google Scholar]
- Jouanguy E, Dupuis S, Pallier A, et al. In a novel form of IFN-gamma receptor 1 deficiency, cell surface receptors fail to bind IFN-gamma, J Clin Invest 2000; 105 : 1429–36. [Google Scholar]
- Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest 1998; 101 : 2364–9. [Google Scholar]
- Fieschi C, Dupuis S, Picard,C, Smith CI, Holland SM, Casanova JL. High levels of interferon gamma in the plasma of children with complete interferon gamma receptor deficiency. Pediatrics 2001; 107 : E48. [Google Scholar]
- Jouanguy E, Lamhamedi-Cherradi S, Altare F, et al. Partial interferon-gamma receptor 1 deficiency in a child with tuberculoid bacillus Calmette-Guerin infection and a sibling with clinical tuberculosis. J Clin Invest 1997; 100 : 2658–64. [Google Scholar]
- Allende LM, Lopez-Goyanes A, Paz-Artal E, et al. A point mutation in a domain of gamma interferon receptor 1 provokes severe immunodeficiency. Clin Diagn Lab Immunol 2001; 8 : 133–7. [Google Scholar]
- Jouanguy E, Lamhamedi-Cherradi S, Lammas D, et al. A human IFNGR1 small deletion hotspot associated with dominant susceptibility to mycobacterial infection. Nat Genet 1999; 21 : 370–8. [Google Scholar]
- Villella A, Picard C, Jouanguy E, et al. Recurrent Mycobacterium avium osteomyelitis associated with a novel dominant interferon gamma receptor mutation. Pediatrics 2001; 107 : E47. [Google Scholar]
- Doffinger R, Jouanguy E, Dupuis S, et al. Partial interferon-gamma receptor signaling chain deficiency in a patient with bacille Calmette-Guerin and Mycobacterium abscessus infection. J Infect Dis 2000; 181 : 379–84. [Google Scholar]
- Dupuis S, Dargemont C, Thomassin N, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science 2001; 293 : 300–3. [Google Scholar]
- Dupuis S, Doffinger R, Picard C, et al. Human interferon-gamma-mediated immunity is a genetically controlled continuous trait that determines the outcome of mycobacterial invasion. Immunol Rev 2000; 178 : 129–37. [Google Scholar]
- Remus N, Reichenbach J, Picard C, et al. Impaired interferon gamma-mediated immunity and susceptibility to mycobacterial infection in childhood. Pediatr Res 2001; 50 : 8–13. [Google Scholar]
- Trinchieri G. Interleukin-12 : a cytokine at the interface of inflammation and immunity. Adv Immunol 1998; 70 : 83–243. [Google Scholar]
- Presky DH, Yang H, Minetti LJ, et al. A functional interleukin 12 receptor complex is composed of two beta- type cytokine receptor subunits. Proc Natl Acad Sci USA 1996; 93 : 14002–7. [Google Scholar]
- Zou J, Presky DH, Wu CY, Gubler U. Differential associations between the cytoplasmic regions of the interleukin-12 receptor subunits beta-1 and beta-2 and JAK kinases. J Biol Chem 1997; 272 : 6073–7. [Google Scholar]
- Jacobson NG, Szabo SJ, Weber-Nordt RM, et al. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J Exp Med 1995; 181 : 1755–62. [Google Scholar]
- Naeger LK, McKinney J, Salvekar A, Hoey,T. Identification of a STAT4 binding site in the interleukin-12 receptor required for signaling. J Biol Chem 1999; 274 : 1875–8. [Google Scholar]
- Barbulescu K, Becker C, Schlaak JF, Schmitt E, Meyer zum Buschenfelde KH, Neurath MF. IL-12 and IL-18 differentially regulate the transcriptional activity of the human IFN-gamma promoter in primary CD4+ T lymphocytes. J Immunol 1998; 160 : 3642–7. [Google Scholar]
- Altare F, Lammas D, Revy P, et al. Inherited interleukin 12 deficiency in a child with bacille Calmette-Guérin infection. J Clin Invest 1998; 102 : 2035–40. [Google Scholar]
- Aksu G, Trpan C, Çavuolu C, Soydan S, Altare F, Casanova JL, Kutukculer N. Mycobacterium fortuitum-chelonae complex infection in a child with complete interleukin-12 receptor beta-1 deficiency. Pediatr Infect Dis J 2001; 20 : 551–3. [Google Scholar]
- Altare F, Durandy A, Lammas D, et al. Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science 1998; 280 : 1432–5. [Google Scholar]
- De Jong R, Altare F, Haagen IA, et al. Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science 1998; 280: 1435–8. [Google Scholar]
- Sakai T, Matsuoka M, Aoki M, Nosaka K, Mitsuya H. Missense mutation of the interleukin-12 receptor beta-1 chain-encoding gene is associated with impaired immunity against Mycobacterium avium complex infection. Blood 2001; 97 : 2688–94. [Google Scholar]
- Altare F, Ensser A, Breiman A, et al. Interleukin-12 receptor β1-deficiency in a patient with abdominal tuberculosis. J Infect Dis 2001; 184 : 231–6. [Google Scholar]
- Altare F, Jouanguy E, Lamhamedi S, Döffinger R, Fischer A, Casanova JL. Mendelian susceptibility to mycobacterial infections in man. Curr Opin Immunol 1998; 10 : 413–7. [Google Scholar]
- Horvath KD, Whelan RL. Intestinal tuberculosis : return of an old disease. Am J Gastroenterol 1998; 93 : 692–6. [Google Scholar]
- Verhagen CE, de Boer T, Smits HH, et al. Residual type 1 immunity in patients genetically deficient for interleukin 12 receptor beta-1 (IL-12R beta-1). Evidence for an IL-12R beta-1-independent pathway of IL-12 responsiveness in human T cells. J Exp Med 2000; 192 : 517–28. [Google Scholar]
- Zhang M, Gately MK, Wang E, et al. Interleukin 12 at the site of disease in tuberculosis, J Clin Invest 1994; 93 : 1733–9. [Google Scholar]
- Zhang M, Gong J, Presky DH, Xue W, Barnes PF. Expression of the IL-12 receptor beta 1 and beta 2 subunits in human tuberculosis. J Immunol 1999; 162 : 2441–7. [Google Scholar]
- Flesch IE, Hess JH, Huang S, et al. Early interleukin 12 production by macrophages in response to mycobacterial infection depends on interferon gamma and tumor necrosis factor alpha. J Exp Med 1995; 181 : 1615–21. [Google Scholar]
- DiTirro J, Rhoades ER, Roberts AD, et al. Disruption of the cellular inflammatory response to Listeria monocytogenes infection in mice with disruptions in targeted genes. Infect Immun 1998; 66 : 2284–9. [Google Scholar]
- Dye C, Scheele S, Dolin P, Pathania V, Raviglione M. Consensus statement. Global burden of tuberculosis : estimated incidence, prevalence and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA 1999; 282 : 677–86. [Google Scholar]
- Abel L, Casanova JL, Genetic predisposition to clinical tuberculosis : bridging the gap between simple and complex inheritance. Am J Hum Genet 2000; 67 : 274–7. [Google Scholar]
Liste des tableaux
Quatre groupes d’individus peuvent être distingués selon la corrélation entre le génotype et le phénotype clinique et cellulaire.
Liste des figures
|
Figure 1. Étiologie génétique du syndrome de prédisposition mendélienne aux infections mycobactériennes. |
| Dans le texte | |
|
Figure 2. Voie de signalisation de l’IFN-γ. |
| Dans le texte | |
|
Figure 3. Hétérogénéité allélique des défauts en IFN-γR1. |
| Dans le texte | |
|
Figure 4. Voie de signalisation de l’IL-12. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.