")
")
| Issue |
Med Sci (Paris)
Volume 40, Number 6-7, Juin-Juillet 2024
|
|
|---|---|---|
| Page(s) | 534 - 543 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2024083 | |
| Published online | 8 juillet 2024 | |
Les phosphodiestérases des nucléotides cycliques
Cibles thérapeutiques dans l’hypertrophie et l’insuffisance cardiaques
Cyclic nucleotide phosphodiesterases: therapeutic targets in cardiac hypertrophy and failure
Université Paris-Saclay, Inserm UMR-S 1180, Orsay, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les phosphodiestérases des nucléotides cycliques (PDE) modulent la régulation neuro-hormonale de la fonction cardiaque en dégradant l’AMPc et le GMPc. Dans les cardiomyocytes, de multiples isoformes de PDE, aux propriétés enzymatiques et aux localisations subcellulaires différentes, régulent localement les niveaux de nucléotides cycliques et les fonctions cellulaires associées. Cette organisation est fortement perturbée au cours de l’hypertrophie et de l’insuffisance cardiaque à fraction d’éjection réduite (IC), ce qui peut contribuer à la progression de la maladie. Sur le plan clinique, l’inhibition des PDE a été considérée comme une approche prometteuse pour compenser la désensibilisation aux catécholamines qui accompagne l’IC. Bien que des inhibiteurs de la PDE3, tels que la milrinone ou l’énoximone, puissent être utilisés cliniquement pour améliorer la fonction systolique et soulager les symptômes de l’IC aiguë, leur utilisation chronique s’est avérée préjudiciable. D’autres PDE, telles que les PDE1, PDE2, PDE4, PDE5, PDE9 et PDE10, sont apparues comme de nouvelles cibles potentielles pour le traitement de l’IC, chacune ayant un rôle unique dans les voies de signalisation locales des nucléotides cycliques. Dans cette revue, nous décrivons la signalisation de l’AMPc et du GMPc dans les cardiomyocytes et présentons les différentes familles de PDE exprimées dans le cœur ainsi que leurs modifications dans l’hypertrophie cardiaque pathologique et dans l’IC. Nous évaluons également les résultats issus de modèles précliniques ainsi que les données cliniques indiquant l’utilisation d’inhibiteurs ou d’activateurs de PDE spécifiques qui pourraient avoir un potentiel thérapeutique dans l’IC.
Abstract
Cyclic nucleotide phosphodiesterases (PDEs) modulate neurohormonal regulation of cardiac function by degrading cAMP and cGMP. In cardiomyocytes, multiple isoforms of PDEs with different enzymatic properties and subcellular locally regulate cyclic nucleotide levels and associated cellular functions. This organisation is severely disrupted during hypertrophy and heart failure (HF), which may contribute to disease progression. Clinically, PDE inhibition has been seen as a promising approach to compensate for the catecholamine desensitisation that accompanies heart failure. Although PDE3 inhibitors such as milrinone or enoximone can be used clinically to improve systolic function and relieve the symptoms of acute CHF, their chronic use has proved detrimental. Other PDEs, such as PDE1, PDE2, PDE4, PDE5, PDE9 and PDE10, have emerged as potential new targets for the treatment of HF, each with a unique role in local cyclic nucleotide signalling pathways. In this review, we describe cAMP and cGMP signalling in cardiomyocytes and present the different families of PDEs expressed in the heart and their modifications in pathological cardiac hypertrophy and HF. We also review results from preclinical models and clinical data indicating the use of specific PDE inhibitors or activators that may have therapeutic potential in CI.
© 2024 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (© Xavier Prieur).
L’insuffisance cardiaque à fraction d’éjection réduite (IC) est un syndrome clinique persistant caractérisé par le déclin progressif de la fonction cardiaque. À terme, la capacité du cœur à pomper efficacement le sang devient insuffisante pour répondre aux besoins métaboliques de l’organisme, ce qui entraîne une défaillance des organes et, dans les cas les plus graves, le décès du patient. Quelle que soit sa cause, la réduction de la fonction cardiaque déclenche l’activation de systèmes neurohormonaux et le développement d’une hypertrophie cardiaque pour normaliser la contrainte exercée sur la paroi ventriculaire [1]. Cependant, cet état compensé est généralement de courte durée et favorise l’évolution vers l’IC.
Dans des conditions physiologiques, la fonction cardiaque est régulée par deux nucléotides cycliques intracellulaires aux effets opposés : l’AMPc et le GMPc. L’AMPc, produit après une stimulation des récepteurs β-adrénergiques (β-AR) en réponse à l’exercice, augmente le débit cardiaque par le biais de la signalisation de la protéine kinase dépendante de l’AMPc (PKA, pour protéine kinase A). Inversement, le GMPc, stimulé par la production d’oxyde nitrique (NO) et par les peptides natriurétiques1 (NP), peut exercer des effets à la fois synergiques et opposés à ceux de l’AMPc sur la fonction cardiaque. Dans l’IC, la diminution de la fonction cardiaque entraîne une suractivation du système nerveux sympathique et une élévation chronique de la concentration de catécholamines [2]. Cela déclenche un « cercle vicieux » physiopathologique, dans lequel la stimulation excessive des β-AR joue un rôle central, comme le montrent les effets bénéfiques des antagonistes des récepteurs β-adrénergiques (ou β-bloquants) dans I’IC [3]. Le maintien de la signalisation AMPc-PKA déclenche un remodelage inadapté, notamment une hypertrophie cardiaque pathologique, la mort des cardiomyocytes et la fibrose cardiaque, ce qui conduit finalement à l’IC. En revanche, le NO et les NP ont des effets anti-hypertrophiques et anti-fibrosants. Par conséquent, les médicaments qui augmentent la production de NP, ou agissent en synergie avec le NO pour stimuler la synthèse du GMPc, améliorent les traitements conventionnels pour les patients souffrant d’insuffisance cardiaque à fraction d’éjection réduite (en anglais, HFrEF pour heart failure with reduced ejection fraction) [4, 5].
Les niveaux de nucléotides cycliques (AMPc et GMPc) sont étroitement contrôlés par leur dégradation par les phosphodiestérases (PDE). La superfamille des PDE se compose d’isoformes distinctes (Figure 1) qui contrôlent l’intensité et la durée des signaux AMPc et GMPc dans des compartiments spécifiques des cardiomyocytes. Des données récentes, datant des cinq dernières années, soulignent ainsi la localisation préférentielle de la dégradation de l’AMPc par les PDE dans des domaines de taille nanométrique (Figure 2) [6, 7].
 |
Figure 1. Structure et propriétés enzymatiques des PDE cardiaques. Les domaines catalytiques conservés dans les régions C-terminales des phosphodiestérases (PDE) sont représentés par des cylindres violets pour les enzymes dégradant à la fois l’AMPc et le GMPc, et sont représentés par des cylindres rouges ou bleus lorsqu’elles hydrolysent spécifiquement l’AMPc ou le GMPc, respectivement. Ces protéines diffèrent par leurs propriétés biochimiques. Pour chaque famille de PDE, les valeurs Km (constante de Michaelis) et Vmax (vitesse maximale) sont indiquées dans les tableaux. Des domaines de ciblage spécifiques et de nombreux types de domaines protéiques assurent le contrôle de l’activité catalytique des PDE et permettent leur localisation subcellulaire. Certains des domaines régulateurs situés dans les régions N-terminales abritent des sites de phosphorylation pour diverses kinases, afin de moduler leurs activités catalytiques. GAFA et GAFB : cGMP-specific and -stimulated phosphodiesterases, Anabaena adenylate cyclases and Escherichia coli FhlA domain A and B ; UCR1 et UCR2 : upstream conserved region 1 and 2 ; LR1 et LR2 : linker region 1 and 2 ; REC : regulator receiver domain ; Pas : per-arnt-sim domain ; Pat7 : signal de localisation nucléaire ; P : résidu phosphorylé ; CAMKII : protéine kinase II Ca2+/calmoduline-dépendante ; ERK : extracellular signal-regulated kinase. |
 |
Figure 2. Localisation subcellulaire et activités des isoformes de PDE au sein des complexes macromoléculaires dans les myocytes cardiaques. Divers signalosomes dans le cardiomyocyte ventriculaire, y compris les phosphodiestérases (PDE), contrôlent des réservoirs différents d’AMPc (fond rouge) synthétisés par l’adénylate cyclase (AC) à la membrane plasmique lorsqu’elle est activée par la protéine G hétérotrimérique stimulant l’adénylate cyclase (Gs), ou produits par les AC solubles (sAC) dans les mitochondries. Les signaux de GMPc (fond bleu) catalysés par les guanylate cyclases particulaires (pGC) et solubles (sGC) sont également illustrés. Diverses isoformes de PDE contrôlent les concentrations de nucléotides cycliques émanant des récepteurs β-adrénergiques (β-AR), des récepteurs de l’adénosine 2A (A2R) et des GC activés par les peptides natriurétiques ou l’oxyde nitrique (NO). Elles sont des modulateurs cruciaux de nombreuses réponses cellulaires à divers stimulus externes, ajustant finement les niveaux d’AMPc et de GMPc dans les domaines subcellulaires pour moduler le couplage excitation-contraction, mais aussi contrôler l’apoptose et l’expression de gènes hypertrophiques en limitant la phosphorylation de facteurs transcriptionnels, tels que la protéine de liaison de l’élément répondant à l’AMPc (CREB pour cAMP response element-binding protein) ou le facteur nucléaire des lymphocytes T activés (NFAT). EPAC1 : facteur d’échange. |
Dans des conditions normales, parallèlement au positionnement stratégique de la PKA au voisinage de ses substrats, facilité par les protéines d’ancrage (AKAP pour A-kinase anchoring proteins), qui compartimentalisent la PKA dans des régions cellulaires précises, les PDE assurent la précision de la signalisation de l’AMPc. Dans l’hypertrophie cardiaque pathologique et l’IC, cette organisation est fortement perturbée, ce qui contribuerait à la détérioration de la fonction cardiaque. En effet, dans l’IC, la signalisation β-AR subit un remodelage important [8], et la biodisponibilité du NO et la signalisation des NP sont altérées [9]. Des modifications de l’expression, de l’activité et de la localisation subcellulaire des PDE sont observées et participent aux altérations de la signalisation de l’AMPc et du GMPc dans l’IC.
À l’heure actuelle, les inhibiteurs de la PDE3 et de la PDE5 ont montré soit des effets néfastes chez les patients atteints d’IC chronique sévère [10], soit une inefficacité chez les patients atteints d’IC à fraction d’éjection préservée (HFpEF pour heart failure with preserved ejection fraction) [11]. Cependant, les recherches en cours suggèrent que certaines PDE spécifiques pourraient être des cibles thérapeutiques potentielles pour prévenir le remodelage cardiaque et l’IC, et les arythmies qui l’accompagnent [12, 13].
Parmi les onze membres principaux de la superfamille des PDE, les PDE1, PDE2, PDE3, PDE4, PDE5, PDE8, PDE9 et PDE10 sont exprimées dans le cœur (Figure 1), et il a été montré qu’elles participentau contrôle de la fonction cardiaque. Le niveau d’expression ou d’activité de certaines PDE est également augmenté dans certaines situations pathologiques, telles que l’hypertrophie cardiaque induite par la surcharge de pression et l’IC. Dans des modèles animaux, l’inhibition pharmacologique de certaines de ces PDE ou l’inactivation des gènes qui les codent se sont avérées cardioprotectrices. D’autres études suggèrent qu’augmenter l’expression et donc l’activité de certaines isoformes pourrait s’avérer cardioprotecteur [14–16]. L’essentiel des travaux et des détails sur la façon dont les PDE influencent le cœur normal et malade, l’hypertrophie cardiaque et la progression vers l’IC sur les PDE chez l’homme, ainsi que les divers essais thérapeutiques ciblant les PDE ont été décrits dans une revue récente [13]. Nous présentons ici les avancées les plus marquantes.
Régulation de la fonction cardiaque par les PDE via la voie de l’AMPc
La PDE1
La PDE1 est la seule PDE connue qui soit stimulée par le complexe calcium-calmoduline (Ca2+/CaM). Son expression dépend de trois gènes codant les isoformes PDE1A, PDE1B et PDE1C. Seules les isoformes PDE1A et PDE1C sont exprimées dans le cœur. La PDE1A domine dans le myocarde de rat et de souris, et cette isoforme semble hydrolyser préférentiellement le GMPc [17]. La PDE1C présente une sélectivité similaire pour le GMPc et pour l’AMPc in vitro, mais dans les myocytes et les cœurs intacts, elle agit principalement sur l’AMPc [18, 19]. Cette isoforme est exprimée de façon plus importante chez les grands mammifères, y compris l’homme [20]. Elle fournit la majeure partie de l’activité basale d’hydrolyse du GMPc et de l’AMPc dans les fractions solubles des myocytes humains [21]. L’expression des PDE1A et PDE1C augmente chez les patients en IC et dans les modèles animaux de cette maladie [17, 19, 22]. La délétion génétique de la PDE1C chez la souris n’entraîne pas de phénotype particulier à l’état basal mais protège contre l’hypertrophie et la fibrose suite à une surcharge de pression, par l’intermédiaire des voies AMPc-PKA et PI3K (phosphoinositide 3-kinase)-AKT (protéine kinase B) [19].
Les effets du blocage pharmacologique chronique de la PDE1 ont été étudiés chez la souris à l’aide d’inhibiteurs non sélectifs de l’isoforme (par exemple l’IC86340), ou de la vinpocétine (inhibiteur plus faible et moins sélectif), dans des études d’une durée de 1 à 2 semaines dans trois situations pathologiques : la cardiopathie causée par une stimulation par l’angiotensine II [22] ; la protéinopathie induite par la mutation R120G (arginine en position 120 de la séquence protéique substituée par une glycine) de la cristalline αB [23] ; et la cardiotoxicité due à la doxorubicine [24]. Dans chacune de ces situations, l’inhibition de la PDE1 a entraîné des effets bénéfiques sur la signalisation AMPc-PKA, l’activation de l’AKT, du protéosome et des voies anti-apoptotiques. Il a également été montré que la PDE1C réside dans un complexe comprenant le récepteur de l’adénosine A2R et le canal calcique TRPC3 (transient receptor potential C3), qui permet l’activation de la PDE1C par le complexe Ca2+/CaM [24]. Le rôle de la PDE1 a été également étudié chez les grands mammifères, où la PDE1C est la forme majoritaire. Chez le chien et le lapin, l’inhibition sélective de la PDE1 (avec l’ITI-214, ou lenrispodun) entraîne une augmentation de la contractilité et de la relaxation, une réduction de la résistance artérielle et une augmentation de la fréquence cardiaque [18]. Le blocage des récepteurs β-AR abolit l’effet chronotrope positif2 du lenrispodun, tandis que le blocage du récepteur A2R-B de l’adénosine abolit les effets inotrope3 et chronotrope positifs, ce qui ajoute un argument supplémentaire en faveur du couplage PDE1-A2R [18]. Des études ultérieures réalisées sur des myocytes isolés ont comparé les mécanismes contractiles couplés à la stimulation des récepteurs β-AR, à l’inhibition de la PDE3 ou à l’inhibition de la PDE1. L’inhibition de la PDE1 augmente l’influx calcique via les canaux calciques voltage dépendants CaV1.2, mais pas la phosphorylation du phospholambane4, de la troponine I ou de la protéine C de liaison à la myosine (MyBPC), ni la charge calcique du réticulum sarcoplasmique (RS)5 [20]. Par rapport à l’inhibition de la PDE3, l’inhibition de la PDE1 conduit à une augmentation plus faible des courants transitoires calciques intracellulaires et, par conséquent, à moins d’arythmies. Ces résultats ont donné lieu au premier et, à ce jour, seul essai clinique d’un inhibiteur de la PDE1 (l’ITI-214) dans l’IC humaine [25].
La PDE2
L’activité et l’expression de la PDE2 sont accrues dans le myocarde de patients souffrant d’IC terminale [16]. Ces augmentations ont également été observées dans le modèle d’IC induite par perfusion chronique d’isoprotérénol chez le rat [16], qui accroît l’hydrolyse de l’AMPc à proximité de la Ca2+-ATPase du RS (ou SERCA2a, pour sarco-endoplasmic reticulum Ca 2+ -ATPase 2a) dans les cardiomyocytes hypertrophiés [26]. L’augmentation de PDE2 entraîne une réduction de la réponse β-AR et protège le cœur du remodelage hypertrophique pathologique induit par la norépinéphrine et la phényléphrine [16]. Ces données suggèrent que l’activation de la PDE2 pourrait être ainsi cardioprotectrice. Cela a en effet été confirmé chez des souris transgéniques surexprimant le gène codant la PDE2 spécifiquement dans le cœur, qui se sont révélées résistantes à la tachycardie ventriculaire induite par les catécholamines, et dont la fonction cardiaque est améliorée après un infarctus du myocarde [15]. Au contraire, l’inhibition de la PDE2, par le BAY 60-7550, a conduit à une plus grande susceptibilité aux arythmies dans les cœurs isolés de souris ex vivo lors d’une reperfusion post-ischémique [27]. À l’inverse, l’augmentation de l’expression de la PDE2 spécifiquement dans le cœur empêche l’apparition de post-dépolarisations induites par l’isoprotérénol [27]. L’ensemble de ces résultats expérimentaux suggère qu’une activation de la PDE2A pourrait constituer une stratégie intéressante pour prévenir l’hypertrophie cardiaque, sa progression vers l’IC, et limiter les arythmies associées.
La PDE2A est une PDE activée par le GMPc [28], et son activité hydrolytique envers l’AMPc peut être multipliée par 30 lorsque le GMPc se lie à son domaine GAFB6. Une augmentation de GMPc, qui est cardioprotectrice dans l’IC [5], pourrait être obtenue en activant la guanylate cyclase particulaire localisée près de la PDE2A [29], et constituer un mécanisme de défense déclenché ainsi par la voie NP/cGMP lors d’un stress cardiaque, en particulier lors d’une activation excessive des récepteurs β-AR. Il est intéressant de noter que le sacubitril, un inhibiteur de l’endopeptidase neutre, qui élève les concentrations de NP, améliore les traitements classiques de l’IC [4], et il a été récemment démontré que le CNP (C-type natriuretic peptide) exerce des effets antiarythmiques via la PDE2 [30].
Alors que ces études fournissent des preuves d’un effet bénéfique de l’activation de la PDE2, d’autres équipes ont rapporté des résultats opposés. La surexpression de la PDE2 s’est en effet avérée pro-hypertrophique [31], et son inhibition par le BAY 60-7550 bloque la croissance des cardiomyocytes via la phosphorylation, par la protéine kinase A (PKA), du facteur nucléaire des lymphocytes T activés (NFAT), empêchant sa translocation vers le noyau et l’activation des gènes pro-hypertrophiques [31]. La PDE2 protège également contre la mort cellulaire apoptotique en favorisant l’élongation mitochondriale par fusion des mitochondries [32]. L’activité et l’expression de la PDE2 sont également augmentées dans les cardiomyocytes hypertrophiés de souris induits par une surcharge de pression et par une stimulation chronique à l’isoprotérénol, agoniste des récepteurs β-AR, et son inhibition par le BAY 60-7550 entraîne une diminution de l’hypertrophie, de la dilatation, de la contractilité et de la fibrose du ventricule gauche [33]. Ces effets bénéfiques du BAY 60 7550 sur la fonction cardiaque pourraient également s’expliquer en partie par l’inhibition de la PDE2 dans le ganglion stellaire, où elle est également augmentée en conditions pathologiques, comme cela a été montré chez des rats hypertendus [34]. La libération accrue de norépinéphrine par les neurones sympathiques aggrave l’hypertension artérielle. L’activité de la PDE2A augmentée dans les neurones, où elle hydrolyse préférentiellement le GMPc, en diminuant l’activité de la protéine kinase G (PKG), contribue à augmenter l’exocytose de la noradrénaline par le système sympathique. En effet, l’élévation de l’activité de la PDE2A constitue un frein à l’action inhibitrice du BNP (brain natriuretic peptide) sur la transmission sympathique, qui, en augmentant le GMPc et ainsi l’activité de la PKG, diminue l’entrée de calcium par le canal CaV1.2. Ainsi, l’inhibition de la PDE2A limiterait la libération de catécholamines, ce qui améliorerait la fonction cardiaque dans ces modèles [34].
À ce jour, il n’y a pas d’explication claire à la divergence dans les résultats publiés concernant la régulation de l’hypertrophie cardiaque par la PDE2, et d’autres études sont nécessaires pour clarifier si l’inhibition ou l’activation de l’enzyme aura des effets thérapeutiques dans l’IC.
La PDE3
La PDE3 est exprimée dans le myocarde de nombreuses espèces, et elle est particulièrement abondante chez les grands mammifères, y compris l’homme. Ainsi, des inhibiteurs de PDE3, tels que la milrinone ou l’enoximone, ont été développés pour le traitement de l’IC, quelle que soit sa cause. Dans l’IC congestive, les inhibiteurs de la PDE3 induisent à court terme une réponse inotrope7 positive et une vasodilatation [35]. Cependant, des études cliniques ont montré qu’à long terme, ces molécules augmentent la mortalité des patients en raison d’arythmies et de morts subites [10]. Bien qu’ils ne soient pas entièrement élucidés, les mécanismes impliqués dans l’effet pro-arythmique des inhibiteurs de PDE3 pourraient inclure une augmentation de la concentration de calcium intracellulaire causée par une augmentation chronique des niveaux d’AMPc [10]. Malgré les effets indésirables des inhibiteurs de PDE3 en clinique, des souris soumises à une constriction de l’aorte thoracique (TAC) et traitées par la milrinone ont montré une diminution du remodelage pathologique du ventricule gauche, de l’hypertrophie et de la fibrose interstitielle [36]. Un effet similaire a été observé chez les souris dont le gène Pde3a a été délété (Pde3a KO) et soumises à une TAC, mais pas chez les souris Pde3b KO, ce qui tend à montrer que les effets bénéfiques de la milrinone observés dans cette étude se produisent via l’inhibition de la PDE3A [36]. Ces travaux suggèrent que les effets indésirables provoqués par l’inhibition de la PDE3 n’impliquent pas la participation de la PDE3A, mais cette question reste controversée [37]. Malgré ces effets positifs de l’inhibition chronique de la PDE3, lors d’expériences d’échocardiographie sur des rats présentant des lésions myocardiques induites par l’isoprotérénol, l’inhibition de la PDE3 par le pimobendan induit un dysfonctionnement diastolique significatif avec une augmentation du dépôt de collagène de type I [38]. L’inhibition de la PDE3 pourrait également être préjudiciable en raison de son effet sur l’apoptose. En effet, in vitro, la PDE3A a été impliquée dans l’activation du répresseur transcriptionnel ICER (inducible cAMP early repressor), qui inhibe l’expression du facteur anti-apoptotique Bcl-2. L’inhibition de la PDE3A entraîne une augmentation des niveaux d’AMPc et une activation conséquente de la PKA, qui augmente ICER, entraînant une diminution de Bcl-2 et l’apoptose des cardiomyocytes [39]. À l’inverse, des souris transgéniques, surexprimant l’isoforme PDE3A1 spécifiquement dans le myocarde, sont protégées dans un modèle d’ischémie-reperfusion, avec une réduction de l’apoptose et de la taille de la zone infarcie. Ces effets bénéfiques ont été associés à une réduction de la voie de signalisation AMPc/PKA/ICER et à une augmentation de la concentration de la protéine Bcl-2. Enfin, il a été montré récemment que l’isoforme PDE3A2 est localisée au niveau nucléaire et réprime l’expression de gènes contrôlant l’hypertrophie cardiaque en limitant l’activité de l’histone déacétylase 1 (HDAC1) [40]. Ces données suggèrent un potentiel thérapeutique de l’activation de la PDE3A1 pour prévenir les effets délétères de l’IC [41]. Il convient de noter que, des mutations dans le gène Pde3a responsables d’une maladie rare caractérisée par la combinaison d’une brachydactylie8 et d’une hypertension, confèrent une cardioprotection malgré l’augmentation de la postcharge [42]. Or, ces mutations entraînent une hyperactivation enzymatique de la PDE3A, ce qui soutient le postulat rapporté précédemment, selon lequel il serait bénéfique d’augmenter l’activité des PDE dégradant l’AMPc dans le tissu cardiaque plutôt que de la diminuer [14].
L’utilisation chronique de l’inhibition de la PDE3 dans l’insuffisance cardiaque reste donc controversée. Dans des modèles d’ischémie reperfusion chez le chien, une injection intraveineuse en bolus d’un inhibiteur de la PDE3, 30 minutes avant l’occlusion coronaire, diminue la taille de la zone infarcie en raison d’une augmentation transitoire des niveaux d’AMPc intracellulaire et de l’activation consécutive de la voie PKA/MAPK (MAP kinase) p38 [43]. Cette activité transitoire de la MAPK p38 est responsable des effets cardioprotecteurs observés dans l’étude. Ces effets sont probablement dus à l’inhibition de la PDE3B, car dans les modèles d’ischémie aiguë in vivo et de cœur perfusé de Langendorff9, et de reperfusion utilisant des souris déficientes pour Pde3a ou Pde3b, la taille de la zone infarcie n’était réduite que chez ces dernières [44].
La PDE4
De nombreuses études précliniques ont montré l’implication de la PDE4 dans la physiopathologie cardiaque. Par exemple, l’amplitude du courant calcique via le canal CaV1.2 sous stimulation β-AR est augmentée chez des souris déficientes pour Pde4b, et ces animaux présentent une augmentation des tachycardies ventriculaires induites par une salve de stimulations électriques, associée à un trouble de la dynamique du calcium intracellulaire [45]. Il a par ailleurs été constaté une diminution de la PDE4B chez les rats présentant une hypertrophie cardiaque induite par une sténose aortique [46], ainsi que chez les patients atteints d’IC [14]. Chez des souris atteintes d’IC, induite par perfusion chronique d’isoprotérénol ou par constriction de l’aorte thoracique, la surexpression de la PDE4B spécifiquement dans le cœur atténue la progression de l’IC [14], ce qui suggère le potentiel thérapeutique du maintien de l’activité de la PDE4B dans le traitement de l’IC. En plus de la PDE4B, les souris déficientes pour Pde4d présentent une augmentation de tachycardies ventriculaires lorsqu’elles sont soumises à un stress physique et développent une cardiomyopathie avec l’âge [47]. Dans le cœur de ces souris, l’absence de PDE4D à proximité des récepteurs de la ryanodine de type 2 (RyR2) conduit à une phosphorylation accrue de ces récepteurs par la PKA, entraînant une fuite accrue de Ca2+ du RS et, par conséquent, des événements arythmogènes [47]. Selon une étude portant sur des patients atteints de cardiomyopathie dilatée idiopathique, l’expression de la PDE4D est réduite dans le myocarde [48]. Dans les cardiomyocytes néonataux, une isoforme particulière de PDE4D, la PDE4D5, forme un complexe avec la β-arrestine 2, et la dissociation de ce complexe conduit à un changement de la signalisation β2-AR non-hypertrophique vers la signalisation β1-AR pro-hypertrophique [49]. La famille des PDE4 a également été impliquée dans le développement de la dysfonction ventriculaire droite induite par surcharge de pression chez le cochon [50]. Dans cette maladie, l’expression de certaines familles de PDE est diminuée, notamment la PDE4, et l’inhibition de la PDE4 provoque des évènements arythmogènes [50]. La PDE4 est également exprimée dans les myocytes d’oreillette humaine, où elle contribue à l’hydrolyse de l’AMPc en contrôlant l’influx de Ca2+ à travers les canaux CaV1.2 dans des conditions basales et sous stimulation β-AR [51]. L’inhibition de PDE4 dans ces cellules atriales entraîne une augmentation de la fréquence des sparks (« étincelles » calciques observées par imagerie confocale, dues à la libération spontanée de Ca2+ du RS) et des vagues de Ca2+, conduisant à des arythmies. En accord avec ces résultats, il a été montré que l’activité de la PDE4 est réduite chez les patients souffrant de fibrillation auriculaire, une condition dans laquelle une altération de la voie de signalisation de l’AMPc et des dynamiques du Ca2+ est observée [51]. Cela souligne l’implication de cette famille de PDE, non seulement au niveau ventriculaire mais aussi au niveau atrial, dans le contrôle des niveaux d’AMPc, à la fois dans des conditions basales et sous stimulation adrénergique, protégeant ainsi contre le développement d’arythmies.
La PDE8
L’importance de la PDE8 dans la physiopathologie cardiaque n’a pas encore été entièrement explorée. Cependant, une étude réalisée sur une souris dont le gène codant l’isoforme PDE8A a été délété a montré une augmentation des courants transitoires calciques et du courant calcique ICa,L ainsi qu’une élévation des sparks de Ca2+ dans les cardiomyocytes ventriculaires traités avec un agoniste des β-AR. À ce jour, il s’agit du seul travail montrant l’implication de cette famille de PDE dans la régulation du couplage excitation-contraction ventriculaire [52]. D’autres travaux plus récents ont montré l’expression des PDE8A et PDE8B dans l’oreillette humaine et ont mis en évidence le rôle de la PDE8B chez les patients atteints de fibrillation auriculaire persistante. Cette isoforme est localisée dans la membrane plasmique et est responsable du contrôle des niveaux d’AMPc dans les myocytes atriaux chez les patients atteints de fibrillation auriculaire paroxystique. L’expression de la PDE8B est par ailleurs augmentée chez ces patients, ce qui contribue à une réduction des niveaux d’AMPc à proximité du canal CaV1.2, conduisant à une diminution du courant ICa,L [53]. Il s’agit de la première démonstration d’un rôle de la PDE8 en tant que régulateur des canaux calciques voltage-dépendants de type L dans les myocytes atriaux humains.
Régulation de la fonction cardiaque par les PDE via la voie du GMPc
PDE5A et PDE9A
Les PDE5A et PDE9A sont toutes deux très sélectives pour l’hydrolyse du GMPc et sont exprimées dans le myocarde à de faibles taux. Dans les cardiomyocytes, elles régulent différents réservoirs intracellulaires de GMPc et la signalisation associée, la PDE5A modulant le GMPc produit par la guanylyl cyclase soluble (GCS) stimulée par le monoxyde d’azote (NO) [54], et la PDE9A modulant le GMPc produit par la guanylate cyclase particulaire GC-A [55]. Cette distinction a des conséquences sur la manière dont l’inhibition sélective de chaque PDE influe sur les modèles de maladies cardiaques. Par exemple, dans les cœurs intacts soumis à une surcharge de pression pathologique, l’inhibition de la PDE5A et de la PDE9A réduit de manière similaire l’hypertrophie et la fibrose, et améliore la fonction cardiaque. Cependant, lorsque les animaux sont traités simultanément par le L-NAME (nitro-L-arginine methyl ester) pour inhiber la NO synthase, seul l’inhibiteur de la PDE9A confère ces effets. Un moyen plus physiologique de réduire la signalisation du NO chez les animaux femelles consiste à retirer leurs ovaires afin de réduire l’activation de la NOS couplée aux œstrogènes. Dans ce cas, le blocage de la PDE5A devient également inefficace pour contrer la surcharge de pression ou d’autres formes de stress pathologique [56]. L’inhibition de la PDE9A reste en revanche efficace chez les femelles ovariectomisées [57]. Les PDE5A et PDE9A sont distribuées différemment dans le myocyte : la PDE5A est présente dans les disques Z délimitant les sarcomères [58], tandis que la PDE9A se trouve dans les tubules transverses et dans les mitochondries [55].
L’expression des PDE5A et PDE9A est augmentée dans l’IC humaine [55, 59], ce qui suggère leur rôle potentiel dans la physiopathologie de l’IC. De nombreuses études testant les effets d’inhibiteurs de PDE5A ou de PDE9A suggèrent en effet ce rôle, montrant une amélioration suite à une cardiopathie ischémique, à une surcharge de pression, à une cardiotoxicité due à la doxorubicine, et autres stress [13].
Alors que de nombreuses études réalisées avec des modèles animaux ont montré des effets bénéfiques d’une inhibition de la PDE5A dans diverses maladies myocardiques, cela n’a pas été confirmé chez l’homme [60]. Aussi, les inhibiteurs de PDE5A sont largement utilisés pour traiter l’hypertension pulmonaire et la dysfonction érectile, alors que les indications dans l’hypertension artérielle restent peu décrites. Dans un modèle ovin d’insuffisance cardiaque aiguë, l’inhibition de la PDE9A, utilisée seule ou en association avec un inhibiteur de la néprilysine (pour augmenter encore les niveaux de GMPc que la PDE9A régulerait à son tour), améliore les fonctions cardiaque et rénale [61]. Les essais cliniques de l’inhibition de PDE9A dans l’IC sont en cours, et il reste à déterminer si les données prometteuses obtenues chez l’animal seront reproduites chez l’homme.
La PDE10A
La PDE10A est une PDE qui hydrolyse à la fois l’AMPc et le GMPc. Son rôle dans le remodelage pathologique du cœur a récemment été découvert [62]. Comme pour les autres PDE (mentionnées ci-dessus), l’expression de la PDE10A augmente lors de l’IC chez la souris et chez l’homme. Dans des cardiomyocytes isolés stimulés par la norépinéphrine, ou dans le cœur intact soumis à une élévation de la postcharge ou à l’angiotensine II, le TP-10, un inhibiteur sélectif de la PDE10A, réduit l’hypertrophie pathologique, la fibrose et la dysfonction myocardique. Les souris présentant une délétion génétique de la PDE10A sont normales au repos, présentent des réponses pathologiques réduites à une surcharge de pression, et ont une survie améliorée. L’inhibition de la PDE10A entraîne une augmentation à la fois de l’AMPc et du GMPc, bien que leur importance relative dans les effets nets sur le cœur reste inconnue. Il a également été constaté que la PDE10A contribuait à la croissance tumorale et aux effets cardiotoxiques de la doxorubicine. En utilisant un modèle de cancer du sein, il a été montré que l’inhibition génétique ou pharmacologique de la PDE10A diminuait les lésions cardiaques résultant de la toxicité de la doxorubicine tout en supprimant simultanément la croissance tumorale [63].
Conclusions
Notre compréhension des mécanismes physiopathologiques de l’insuffisance cardiaque et des arythmies associées a beaucoup progressé. Des études réalisées avec des modèles animaux d’IC ont montré que les PDE sont des cibles thérapeutiques potentielles. Cependant, les mécanismes cellulaires contrôlés par chaque famille ou isoforme de PDE sont complexes et soulignent la nécessité de cibler des isoformes spécifiques de PDE dans des microdomaines circonscrits des cellules myocardiques, plutôt que d’utiliser une approche « pan-PDE » pour produire des changements globaux dans les concentrations de nucléotides cycliques [64].
Plusieurs inhibiteurs de PDE ont été testés dans le cadre d’essais cliniques pour leurs effets cardioprotecteurs potentiels chez les patients atteints d’IC mais, malheureusement, malgré des observations préliminaires encourageantes, aucun bénéfice clair n’a finalement été rapporté [13]. Plusieurs facteurs peuvent expliquer l’échec de la transposition des résultats prometteurs des études précliniques dans l’arène clinique : premièrement, les différences de patrons d’expression des familles et isoformes de PDE entre espèces animales ; deuxièmement, dans la plupart des études précliniques, la modulation d’activité de PDE intervient avant (modèles transgéniques ou knock-out) ou en même temps (traitement pharmacologique) que l’apparition de l’IC, ce qui favorise la prévention de la maladie. Or dans les études cliniques, sont traités les patients souffrant d’une IC établie, un scénario qui pourrait nécessiter des modulations différentes de PDE ; troisièmement, même des modulateurs sélectifs d’isoformes particulières de PDE peuvent être inefficaces lorsque l’isoforme est exprimée dans plusieurs compartiments, en particulier lorsque ces compartiments contrôlent des fonctions opposées. La mise au point de modulateurs sélectifs d’une isoforme de PDE particulière dans le compartiment d’intérêt, par exemple en utilisant un inhibiteur peptidique capable de pénétrer dans les cellules pour déplacer l’isoforme de PDE de son complexe de signalisation [65], pourrait contribuer à résoudre ce problème ; enfin, l’interaction complexe entre les voies de signalisation intracellulaire de l’AMPc et du GMPc (Figure 3) et la présence de diverses isoformes de PDE dans différents compartiments pourraient nécessiter une polythérapie pour ajuster avec précision les niveaux de nucléotides cycliques dans des compartiments subcellulaires spécifiques. Par exemple, l’activation de la PDE4D près du récepteur de la ryanodine RYR2 (ryanodine receptor 2), ou de la PDE4B ou de la PDE2A près des canaux calciques voltage dépendants CaV1.2, tout en inhibant la PDE3A ou la PDE5 près du phospholambane, pourrait rétablir une compartimentation normale de l’AMPc et du GMPc dans l’hypertrophie et prévenir la transition vers l’IC. Des études complémentaires portant sur ces questions sont nécessaires pour déterminer si les modulateurs de PDE peuvent être appliqués avec succès pour traiter les patients souffrant d’hypertrophie cardiaque et d’IC.
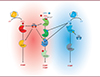 |
Figure 3. Les enzymes impliquées dans la synthèse et la dégradation de l’AMPc et du GMPc dans les cardiomyocytes. Les adénylate cyclases (AC) et les guanylate cyclases (GC) sont impliquées dans la synthèse de l’AMPc (en rouge) et du GMPc (en bleu), respectivement, et diverses phosphodiestérases (PDE) dégradent les deux nucléotides cycliques dans les cardiomyocytes. Les PDE sont à l’interface entre les deux voies de signalisation dans les myocytes cardiaques. Les PDE1, PDE2, PDE3 et PDE10 hydrolysent à la fois l’AMPc et le GMPc, tandis que les PDE4 et PDE5 sont spécifiques de l’AMPc ou du GMPc, respectivement. L’implication des PDE dans les interactions complexes entre ces voies de signalisation est favorisée par leurs différentes propriétés enzymatiques et leur régulation directe par les nucléotides cycliques : par exemple, l’activation de la PDE2 et de la PDE5 par le GMPc, la dégradation limitée de l’AMPc par la PDE3 lorsque le GMPc se lie à sa région catalytique et l’inhibition de la PDE10 par l’AMPc. Ainsi, en fonction de leur localisation subcellulaire, les PDE peuvent modifier les activités enzymatiques d’autres PDE en dégradant l’un ou l’autre des nucléotides cycliques. En outre, certaines PDE sont phosphorylées par des kinases, telles que la sous-unité catalytique de la protéine kinase dépendante de l’AMPc (PKA), qui active les PDE1, PDE3, PDE4 et PDE8, ou par la protéine kinase dépendante du GMPc (PKG), qui augmente l’activité hydrolytique de la PDE5, fournissant des boucles de rétroaction négative pour façonner les signaux de l’AMPc et du GMPc au sein du cardiomyocyte. (P) : phosphorylation. |
Liens d’intérêts
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Le travail réalisé dans notre laboratoire a été financé par the Leducq Foundation for Cardiovascular Research (19CVD02), ERA-CVD “PDE4HEART” ANR-16-ECVD-0007-01, et ANR-23-CE14-0027-01 à RF, ANR-19-CE14-0038-02 et ANR-21-CE14-0082-01 à GV, ANR-23-CE14-0051-01 à JL. Rima Kamel était financée par une bourse post-doctorale de ERA-CVD et de la Fondation Lefoulon-Delalande.
Un peptide natriurétique est un peptide qui induit la natriurèse, c’est-à-dire l’excrétion de sodium par les reins (d’après Wikipédia, ndlr).
Accélération de la fréquence cardiaque.
Augmentation de la force des contractions.
Le phospholambane régule négativement les pompes ATPases calciques (Ca2+- ATPase) du réticulum sarcoplasmique (ndlr).
Le réticulum sarcoplasmique est un réseau de cavités constituant un compartiment de la cellule musculaire dans lequel le calcium est mis en réserve (ndlr).
Le GMPc se lie à la PDE2 au niveau du domaine GAFB de l’enzyme (le sigle GAF reprend les initiales des trois protéines dans lesquelles ce domaine a été identifié) (ndlr).
Relatif à la force des contractions musculaires et notamment cardiaques (ndlr).
Raccourcissement des doigts et des orteils (ndlr).
Le modèle de Langendorff est un modèle de cœur ex vivo qui permet l’analyse des effets cardiaques inotropes, chronotropes et vasculaires (ndlr).
Références
- Hartupee J, Mann DL. Neurohormonal activation in heart failure with reduced ejection fraction. Nat Rev Cardiol 2017 ; 14 : 30–38. [CrossRef] [PubMed] [Google Scholar]
- Cohn JN, Levine TB, Olivari MT, et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med 1984 ; 311 : 819–823. [CrossRef] [PubMed] [Google Scholar]
- El-Armouche A, Eschenhagen T. Beta-adrenergic stimulation and myocardial function in the failing heart. Heart Fail Rev 2009 ; 14 : 225–241. [CrossRef] [PubMed] [Google Scholar]
- McMurray JJ, Packer M, Desai AS, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014 ; 371 : 993–1004. [CrossRef] [PubMed] [Google Scholar]
- Petraina A, Nogales C, Krahn T, et al. Cyclic GMP modulating drugs in cardiovascular diseases: Mechanism-based network pharmacology. Cardiovasc Res 2022; 118 : 2085–102. [CrossRef] [PubMed] [Google Scholar]
- Anton SE, Kayser C, Maiellaro I, et al. Receptor-associated independent cAMP nanodomains mediate spatiotemporal specificity of GPCR signaling. Cell 2022; 185 : 1130–42. [CrossRef] [PubMed] [Google Scholar]
- Bock A, Annibale P, Konrad C, et al. Optical mapping of cAMP signaling at the nanometer scale. Cell 2020; 182 : 1519–30. [CrossRef] [PubMed] [Google Scholar]
- Nikolaev VO, Moshkov A, Lyon AR, et al. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010 ; 327 : 1653–1657. [CrossRef] [PubMed] [Google Scholar]
- Dickey DM, Dries DL, Margulies KB, Potter LR. Guanylyl cyclase (GC)-A and GC-B activities in ventricles and cardiomyocytes from failed and non-failed human hearts: GC-A is inactive in the failed cardiomyocyte. J Mol Cell Cardiol 2012 ; 52 : 727–732. [CrossRef] [PubMed] [Google Scholar]
- Packer M, Carver JR, Rodeheffer RJ, et al. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N Engl J Med 1991 ; 325 : 1468–1475. [CrossRef] [PubMed] [Google Scholar]
- Redfield MM, Chen HH, Borlaug BA, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 2013 ; 309 : 1268–1277. [CrossRef] [PubMed] [Google Scholar]
- Chen S, Yan C. An update of cyclic nucleotide phosphodiesterase as a target for cardiac diseases. Expert Opin Drug Discov 2021; 16 : 183–96. [CrossRef] [PubMed] [Google Scholar]
- Kamel R, Leroy J, Vandecasteele G, Fischmeister R. Phosphodiesterases as therapeutic targets in cardiac hypertrophy and heart failure. Nat Rev Cardiol 2023; 20 : 90–108. [CrossRef] [PubMed] [Google Scholar]
- Karam S, Margaria JP, Bourcier A, et al. Cardiac overexpression of PDE4B blunts b-adrenergic response and maladaptive remodeling in heart failure. Circulation 2020; 142 : 161–74. [CrossRef] [PubMed] [Google Scholar]
- Vettel C, Lindner M, Dewenter M, et al. Phosphodiesterase 2 protects against catecholamine-induced arrhythmias and preserves contractile function after myocardial infarction. Circ Res 2017 ; 120 : 120–132. [CrossRef] [PubMed] [Google Scholar]
- Mehel H, Emons J, Vettel C, et al. Phoshodiesterase-2 is upregulated in human failing hearts and blunts ß-adrenergic responses in cardiomyocytes. J Am Coll Cardiol 2013 ; 62 : 1596–1606. [CrossRef] [PubMed] [Google Scholar]
- Miller CL, Oikawa M, Cai Y, et al. Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ Res 2009 ; 105 : 956–964. [CrossRef] [PubMed] [Google Scholar]
- Hashimoto T, Kim GE, Tunin RS, et al. Acute enhancement of cardiac function by phosphodiesterase type 1 inhibition - A translational study in the dog and rabbit. Circulation 2018 ; 138 : 1974–1987. [CrossRef] [PubMed] [Google Scholar]
- Knight W, Chen S, Zhang Y, et al. PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc Natl Acad Sci USA 2016 ; 113 : E7116–E7E25. [CrossRef] [PubMed] [Google Scholar]
- Muller GK, Song J, Jani V, et al. PDE1 inhibition modulates Cav1.2 channel to stimulate cardiomyocyte contraction. Circ Res 2021; 129 : 872–86. [CrossRef] [PubMed] [Google Scholar]
- Vandeput F, Wolda SL, Krall J, et al. Cyclic nucleotide phosphodiesterase PDE1C1in human cardiac myocytes. J Biol Chem 2007 ; 282 : 32749–32757. [CrossRef] [PubMed] [Google Scholar]
- Wu MP, Zhang YS, Xu X, et al. Vinpocetine attenuates pathological cardiac remodeling by inhibiting cardiac hypertrophy and fibrosis. Cardiovasc Drugs Ther 2017 ; 31 : 157–166. [CrossRef] [PubMed] [Google Scholar]
- Zhang H, Pan B, Wu P, et al. PDE1 inhibition facilitates proteasomal degradation of misfolded proteins and protects against cardiac proteinopathy. Sci Adv 2019; 5 : eaaw5870. [CrossRef] [PubMed] [Google Scholar]
- Zhang Y, Knight W, Chen S, et al. Multiprotein complex with TRPC (transient receptor potential-canonical) channel, PDE1C (phosphodiesterase 1C), and A2R (adenosine A2 receptor) plays a critical role in regulating cardiomyocyte cAMP and survival. Circulation 2018 ; 138 : 1988–2002. [CrossRef] [PubMed] [Google Scholar]
- Gilotra NA, DeVore AD, Povsic TJ, et al. Acute hemodynamic effects and tolerability of phosphodiesterase-1 inhibition with ITI-214 in human systolic heart failure. Circ Heart Fail 2021; 14 : e008236. [CrossRef] [PubMed] [Google Scholar]
- Sprenger JU, Perera RK, Steinbrecher JH, et al. In vivo model with targeted cAMP biosensor reveals changes in receptor-microdomain communication in cardiac disease. Nat Commun 2015 ; 6 : 6965. [CrossRef] [PubMed] [Google Scholar]
- Wagner M, Sadek MS, Dybkova N, et al. Cellular mechanisms of the anti-arrhythmic effect of cardiac PDE2 overexpression. Int J Mol Sci 2021; 22 : 4816. [CrossRef] [PubMed] [Google Scholar]
- Martins TJ, Mumby MC, Beavo JA. Purification and characterization of a cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from bovine tissues. J Biol Chem 1982 ; 257 : 1973–1979. [CrossRef] [PubMed] [Google Scholar]
- Castro LR, Verde I, Cooper DMF, Fischmeister R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation 2006 ; 113 : 2221–2228. [CrossRef] [PubMed] [Google Scholar]
- Cachorro E, Gunscht M, Schubert M, et al. CNP promotes antiarrhythmic effects via phosphodiesterase 2. Circ Res 2023; 132 : 400–14. [CrossRef] [PubMed] [Google Scholar]
- Zoccarato A, Surdo NC, Aronsen JM, et al. Cardiac hypertrophy is inhibited by a local pool of cAMP regulated by phosphodiesterase 2. Circ Res 2015 ; 117 : 707–719. [CrossRef] [PubMed] [Google Scholar]
- Monterisi S, Lobo MJ, Livie C, et al. PDE2A2 regulates mitochondria morphology and apoptotic cell death via local modulation of cAMP/PKA signalling. Elife 2017 ; 6 : e21374. [CrossRef] [PubMed] [Google Scholar]
- Baliga RS, Preedy MEJ, Dukinfield MS, et al. Phosphodiesterase 2 inhibition preferentially promotes NO/guanylyl cyclase/cGMP signaling to reverse the development of heart failure. Proc Natl Acad Sci USA 2018 ; 115 : E7428–E7E37. [CrossRef] [PubMed] [Google Scholar]
- Liu K, Li D, Hao G, et al. Phosphodiesterase 2A as a therapeutic target to restore cardiac neurotransmission during sympathetic hyperactivity. JCI Insight 2018; 3 : pii: 98694. [CrossRef] [PubMed] [Google Scholar]
- Movsesian M, Wever-Pinzon O, Vandeput F. PDE3 inhibition in dilated cardiomyopathy. Curr Opin Pharmacol 2011 ; 11 : 707–713. [CrossRef] [PubMed] [Google Scholar]
- Polidovitch N, Yang S, Sun H, et al. Phosphodiesterase type 3A (PDE3A), but not type 3B (PDE3B), contributes to the adverse cardiac remodeling induced by pressure overload. J Mol Cell Cardiol 2019 ; 132 : 60–70. [CrossRef] [PubMed] [Google Scholar]
- Movsesian MA. PDE3 inhibition in dilated cardiomyopathy: reasons to reconsider. J Card Fail 2003 ; 9 : 475–480. [CrossRef] [PubMed] [Google Scholar]
- Nakata TM, Suzuki K, Uemura A, et al. Contrasting effects of inhibition of phosphodiesterase 3 and 5 on cardiac function and interstitial fibrosis in rats with isoproterenol-induced cardiac dysfunction. Jf Cardiovasc Pharmacol 2019 ; 73 : 195–205. [CrossRef] [PubMed] [Google Scholar]
- Yan C, Miller CL, Abe J. Regulation of phosphodiesterase 3 and inducible cAMP early repressor in the heart. Circ Res 2007 ; 100 : 489–501. [CrossRef] [PubMed] [Google Scholar]
- Subramaniam G, Schleicher K, Kovanich D, et al. Integrated proteomics unveils nuclear PDE3A2 as a regulator of cardiac myocyte hypertrophy. Circ Res 2023; 132 : 828–48. [CrossRef] [PubMed] [Google Scholar]
- Oikawa M, Wu M, Lim S, et al. Cyclic nucleotide phosphodiesterase 3A1 protects the heart against ischemia-reperfusion injury. J Mol Cell Cardiol 2013 ; 64 : 11–19. [CrossRef] [PubMed] [Google Scholar]
- Ercu M, Mucke MB, Pallien T, et al. Mutant phosphodiesterase 3A protects from hypertension-induced cardiac damage. Circulation 2022; 146 : 1758–78. [CrossRef] [PubMed] [Google Scholar]
- Sanada S, Kitakaze M, Papst PJ, et al. Cardioprotective effect afforded by transient exposure to phosphodiesterase III inhibitors -The role of protein kinase A and p38 mitogen-activated protein kinase. Circulation 2001 ; 104 : 705–710. [CrossRef] [PubMed] [Google Scholar]
- Chung YW, Lagranha C, Chen Y, et al. Targeted disruption of PDE3B, but not PDE3A, protects murine heart from ischemia/reperfusion injury. Proc Natl Acad Sci USA 2015 ; 112 : E2253–E2262. [Google Scholar]
- Leroy J, Richter W, Mika D, et al. Phosphodiesterase 4B in the cardiac L-type Ca2+ channel complex regulates Ca2+ current and protects against ventricular arrhythmias. J Clin Invest 2011 ; 121 : 2651–2661. [CrossRef] [PubMed] [Google Scholar]
- Abi-Gerges A, Richter W, Lefebvre F, et al. Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals. Circ Res 2009 ; 105 : 784–792. [CrossRef] [PubMed] [Google Scholar]
- Lehnart SE, Wehrens XHT, Reiken S, et al. Phosphodiesterase 4D deficiency in the ryanodine receptor complex promotes heart failure and arrhythmias. Cell 2005 ; 123 : 23–35. [Google Scholar]
- Richter W, Xie M, Scheitrum C, et al. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res Cardiol 2011 ; 106 : 249–262. [CrossRef] [PubMed] [Google Scholar]
- Berthouze-Duquesnes M, Lucas A, Sauliere A, et al. Specific interactions between Epac1, β-arrestin2 and PDE4D5 regulate β-adrenergic receptor subtypes differential effects on cardiac hypertrophic signaling. Cell Signal 2013 ; 25 : 970–980. [CrossRef] [PubMed] [Google Scholar]
- Mika D, Bobin P, Lindner M, et al. Synergic PDE3 and PDE4 control intracellular cAMP and cardiac excitation-contraction coupling in a porcine model. J Mol Cell Cardiol 2019 ; 133 : 57–66. [CrossRef] [PubMed] [Google Scholar]
- Molina CE, Leroy J, Xie M, et al. Cyclic adenosine monophosphate phosphodiesterase type 4 protects against atrial arrhythmias. J Am Coll Cardiol 2012 ; 59 : 2182–2190. [CrossRef] [PubMed] [Google Scholar]
- Patrucco E, Albergine MS, Santana LF, Beavo JA. Phosphodiesterase 8A (PDE8A) regulates excitation-contraction coupling in ventricular myocytes. J Mol Cell Cardiol 2010 ; 49 : 330–333. [CrossRef] [PubMed] [Google Scholar]
- Grammatika Pavlidou N, Dobrev S, Beneke K, et al. Phosphodiesterase 8 governs cAMP/PKA-dependent reduction of L-type calcium current in human atrial fibrillation: a novel arrhythmogenic mechanism. Eur Heart J 2023 : 2483–94. [CrossRef] [PubMed] [Google Scholar]
- Takimoto E, Champion HC, Li M, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med 2005 ; 11 : 214–222. [CrossRef] [PubMed] [Google Scholar]
- Lee DI, Zhu G, Sasaki T, et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015 ; 519 : 472–476. [CrossRef] [PubMed] [Google Scholar]
- Fukuma N, Takimoto E, Ueda K, et al. Estrogen receptor-a non-nuclear signaling confers cardioprotection and is essential to cGMP-PDE5 inhibition efficacy. JACC Basic Transl Sci 2020; 5 : 282–95. [CrossRef] [PubMed] [Google Scholar]
- Mishra S, Sadagopan N, Dunkerly-Eyring B, et al. Inhibition of phosphodiesterase type 9 reduces obesity and cardiometabolic syndrome in mice. J Clin Invest 2021; 131 : e148798. [CrossRef] [PubMed] [Google Scholar]
- Nagayama T, Zhang M, Hsu S, et al. Sustained soluble guanylate cyclase stimulation offsets nitric-oxide synthase inhibition to restore acute cardiac modulation by sildenafil. J Pharmacol Exp Ther 2008 ; 326 : 380–387. [CrossRef] [PubMed] [Google Scholar]
- Pokreisz P, Vandenwijngaert S, Bito V, et al. Ventricular phosphodiesterase-5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation 2009 ; 119 : 408–416. [CrossRef] [PubMed] [Google Scholar]
- Cooper TJ, Cleland JGF, Guazzi M, et al. Effects of sildenafil on symptoms and exercise capacity for heart failure with reduced ejection fraction and pulmonary hypertension (the SilHF study): a randomized placebo-controlled multicentre trial. Eur J Heart Fail 2022; 24 : 1239–48. [CrossRef] [PubMed] [Google Scholar]
- Scott NJA, Prickett TCR, Charles CJ, et al. Augmentation of natriuretic peptide bioactivity via combined inhibition of neprilysin and phosphodiesterase-9 in heart failure. JACC Heart Fail 2023; 11 : 227–39. [CrossRef] [PubMed] [Google Scholar]
- Chen S, Zhang Y, Lighthouse JK, et al. A novel role of cyclic nucleotide phosphodiesterase 10A in pathological cardiac remodeling and dysfunction. Circulation 2020; 141 : 217–33. [CrossRef] [PubMed] [Google Scholar]
- Chen S, Chen J, Du W, et al. PDE10A inactivation prevents doxorubicin-induced cardiotoxicity and tumor growth. Circ Res 2023; 133 : 138–57. [CrossRef] [PubMed] [Google Scholar]
- Baillie GS, Tejeda GS, Kelly MP. Therapeutic targeting of 3’,5’-cyclic nucleotide phosphodiesterases: inhibition and beyond. Nat Rev Drug Discov 2019 ; 18 : 770–796. [CrossRef] [PubMed] [Google Scholar]
- Blair CM, Baillie GS. Reshaping cAMP nanodomains through targeted disruption of compartmentalised phosphodiesterase signalosomes. Biochem Soc Trans 2019 ; 47 : 1405–1414. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Structure et propriétés enzymatiques des PDE cardiaques. Les domaines catalytiques conservés dans les régions C-terminales des phosphodiestérases (PDE) sont représentés par des cylindres violets pour les enzymes dégradant à la fois l’AMPc et le GMPc, et sont représentés par des cylindres rouges ou bleus lorsqu’elles hydrolysent spécifiquement l’AMPc ou le GMPc, respectivement. Ces protéines diffèrent par leurs propriétés biochimiques. Pour chaque famille de PDE, les valeurs Km (constante de Michaelis) et Vmax (vitesse maximale) sont indiquées dans les tableaux. Des domaines de ciblage spécifiques et de nombreux types de domaines protéiques assurent le contrôle de l’activité catalytique des PDE et permettent leur localisation subcellulaire. Certains des domaines régulateurs situés dans les régions N-terminales abritent des sites de phosphorylation pour diverses kinases, afin de moduler leurs activités catalytiques. GAFA et GAFB : cGMP-specific and -stimulated phosphodiesterases, Anabaena adenylate cyclases and Escherichia coli FhlA domain A and B ; UCR1 et UCR2 : upstream conserved region 1 and 2 ; LR1 et LR2 : linker region 1 and 2 ; REC : regulator receiver domain ; Pas : per-arnt-sim domain ; Pat7 : signal de localisation nucléaire ; P : résidu phosphorylé ; CAMKII : protéine kinase II Ca2+/calmoduline-dépendante ; ERK : extracellular signal-regulated kinase. |
| Dans le texte | |
|
Figure 2. Localisation subcellulaire et activités des isoformes de PDE au sein des complexes macromoléculaires dans les myocytes cardiaques. Divers signalosomes dans le cardiomyocyte ventriculaire, y compris les phosphodiestérases (PDE), contrôlent des réservoirs différents d’AMPc (fond rouge) synthétisés par l’adénylate cyclase (AC) à la membrane plasmique lorsqu’elle est activée par la protéine G hétérotrimérique stimulant l’adénylate cyclase (Gs), ou produits par les AC solubles (sAC) dans les mitochondries. Les signaux de GMPc (fond bleu) catalysés par les guanylate cyclases particulaires (pGC) et solubles (sGC) sont également illustrés. Diverses isoformes de PDE contrôlent les concentrations de nucléotides cycliques émanant des récepteurs β-adrénergiques (β-AR), des récepteurs de l’adénosine 2A (A2R) et des GC activés par les peptides natriurétiques ou l’oxyde nitrique (NO). Elles sont des modulateurs cruciaux de nombreuses réponses cellulaires à divers stimulus externes, ajustant finement les niveaux d’AMPc et de GMPc dans les domaines subcellulaires pour moduler le couplage excitation-contraction, mais aussi contrôler l’apoptose et l’expression de gènes hypertrophiques en limitant la phosphorylation de facteurs transcriptionnels, tels que la protéine de liaison de l’élément répondant à l’AMPc (CREB pour cAMP response element-binding protein) ou le facteur nucléaire des lymphocytes T activés (NFAT). EPAC1 : facteur d’échange. |
| Dans le texte | |
|
Figure 3. Les enzymes impliquées dans la synthèse et la dégradation de l’AMPc et du GMPc dans les cardiomyocytes. Les adénylate cyclases (AC) et les guanylate cyclases (GC) sont impliquées dans la synthèse de l’AMPc (en rouge) et du GMPc (en bleu), respectivement, et diverses phosphodiestérases (PDE) dégradent les deux nucléotides cycliques dans les cardiomyocytes. Les PDE sont à l’interface entre les deux voies de signalisation dans les myocytes cardiaques. Les PDE1, PDE2, PDE3 et PDE10 hydrolysent à la fois l’AMPc et le GMPc, tandis que les PDE4 et PDE5 sont spécifiques de l’AMPc ou du GMPc, respectivement. L’implication des PDE dans les interactions complexes entre ces voies de signalisation est favorisée par leurs différentes propriétés enzymatiques et leur régulation directe par les nucléotides cycliques : par exemple, l’activation de la PDE2 et de la PDE5 par le GMPc, la dégradation limitée de l’AMPc par la PDE3 lorsque le GMPc se lie à sa région catalytique et l’inhibition de la PDE10 par l’AMPc. Ainsi, en fonction de leur localisation subcellulaire, les PDE peuvent modifier les activités enzymatiques d’autres PDE en dégradant l’un ou l’autre des nucléotides cycliques. En outre, certaines PDE sont phosphorylées par des kinases, telles que la sous-unité catalytique de la protéine kinase dépendante de l’AMPc (PKA), qui active les PDE1, PDE3, PDE4 et PDE8, ou par la protéine kinase dépendante du GMPc (PKG), qui augmente l’activité hydrolytique de la PDE5, fournissant des boucles de rétroaction négative pour façonner les signaux de l’AMPc et du GMPc au sein du cardiomyocyte. (P) : phosphorylation. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.