")
")
| Issue |
Med Sci (Paris)
Volume 40, Novembre 2024
Les Cahiers de Myologie
|
|
|---|---|---|
| Page(s) | 30 - 33 | |
| Section | Prix SFM | |
| DOI | https://doi.org/10.1051/medsci/2024135 | |
| Published online | 18 November 2024 | |
Du gène à la cellule
Validation fonctionnelle des variants RYR1
From gene to cell: Functional validation of RYR1 variants
1
Université Grenoble Alpes, Inserm U1216, Grenoble Institut Neurosciences, Grenoble, France
2
CHU Grenoble Alpes, Grenoble, France
3
Université Grenoble-Alpes, Département de Pharmacochimie Moléculaire (DPM), CNRS UMR 5063, Grenoble, France
4
Laboratoire des Techniques de l’Ingénierie Médicale et de la Complexité – Informatique, Mathématiques et Applications, UMR 5525, Grenoble, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Le dépistage génétique des maladies rares permet d’identifier le(s) gène(s) responsable(s) chez environ 50 % des patients. Les cas restants se trouvent dans une impasse diagnostique, car les connaissances actuelles ne permettent pas d’identifier le bon gène ou de déterminer si le(s) variant(s) détecté(s) sur le gène est(sont) pathogène(s) ou bénin(s). On parle alors de « variants de signification inconnue » (VSI). Dans le cas des maladies neuromusculaires, le gène RYR1 est souvent mis en cause, mais la majorité de ses variants identifiés sont classés comme VSI, ce qui met à mal le diagnostic précis des patients. Notre projet vise à créer un pipeline d’analyses en combinant différentes approches (l’intelligence artificielle, les données de biologies structurales et les analyses fonctionnelles), afin d’obtenir une classification des variants de RYR1 plus efficace et d’améliorer le diagnostic génétique des maladies liées à ce gène.
Abstract
Genetic screening of rare diseases allows identification of the responsible gene(s) in about 50% of patients. The remaining cases are in a diagnostic deadlock as current knowledge fails to identify the correct gene or determine if the detected variant on the gene is pathogenic. These are named “variants of unknown significance” (VUS). In the case of neuromuscular diseases, the RYR1 gene is often implicated, with the majority of variants classified as VUS, requiring reliable classification to help patient diagnosis. Our project aims to create an efficient classification pipeline, integrating artificial intelligence, structural biology data, and functional analyses to enhance genetic diagnosis of RYR1-related diseases.
© 2024 médecine/sciences – Inserm

© J. Rendu
Contexte scientifique de l’étude
Le gène RYR1, situé sur le chromosome 19 du génome humain, est un gène d’environ 150 kb, comprenant 106 exons, codant une protéine de 5 038 acides aminés (~565 kDa) appelée le récepteur de la ryanodine de type 1 (RyR1). Cette protéine forme un homotétramère de plus de 2 MDa dont la fonction principale est la libération du calcium (Ca2+) du réticulum sarcoplasmique (RS) vers le cytoplasme, suite à la propagation d’un potentiel d’action généré au niveau de la terminaison axonale d’un motoneurone. Cette imposante protéine est structurée en une large partie cytosolique en N-terminal et un domaine C-terminal organisé en six hélices transmembranaires formant le pore du canal et ancrant la protéine dans la membrane du RS. Les parties N-terminale et C-terminale contiennent des sites de liaison pour de nombreuses protéines/molécules régulatrices telles que la triadine [1] (Figure 1).
 |
Figure 1 Le complexe de relargage calcique (CRC) des cellules musculaires. Représentation schématique du complexe de relargage calcique (CRC) à la triade. La protéine RyR1 est ancrée dans la membrane du réticulum sarcoplasmique (RS) et libère le calcium (Ca2+) dans le cytoplasme, en réponse à un potentiel d’action produit à la terminaison axonale. L’augmentation du Ca2+ intracellulaire induit le glissement de la myosine sur les filaments d’actine et la contraction musculaire. Des variations sur le gène RYR1 peuvent conduire à un défaut de libération du Ca2+ et à une perturbation du couplage excitation/contraction. |
Sur les 150 kb que compte le gène RYR1, plus de 8 000 mutations ont été identifiées (base de données ClinVar [2], avril 2024). Elles peuvent prendre plusieurs formes : duplication, modification, délétion ou insertion d’un ou plusieurs nucléotides. Ces modifications peuvent avoir des conséquences moléculaires telles qu’un décalage du cadre de lecture de l’ADN, des mutations faux-sens qui modifient des acides aminés, des mutations non-sens qui créent des codons « stop » ou des défauts d’épissage de l’ARN. Ces mutations peuvent perturber la régulation du canal, entraînant des anomalies de l’homéostasie calcique. On regroupe les conséquences fonctionnelles d’une mutation du gène RYR1 dans trois principales catégories : hyperactivité du canal, diminution de la fonction du canal et réduction de la quantité de protéine RyR1.
Ces anomalies sont associées à des présentations histologiques et cliniques variées, avec un degré de sévérité variable. Le terme de « myopathies liées au gène RYR1 » inclut plusieurs entités parmi lesquelles les myopathies à cores centraux, les myopathies centronucléaires, les myopathies à multi-minicores ou à « dusty core ». Au-delà des myopathies, des mutations du gène RYR1 ont également été identifiées chez des patients ayant présenté une hyperthermie maligne peranesthésique ou une hyperthermie d’effort [3].
L’identification des mutations est l’une des étapes majeures du diagnostic. Cela permet d’établir une relation entre le génotype et le phénotype et d’adapter la prise en charge des malades. Le développement croissant des techniques de séquençage de nouvelle génération a ainsi permis d’identifier plus de 8 000 variants du gène RYR1. Depuis quelques années, l’obstacle majeur au diagnostic n’est donc plus le séquençage de l’ADN des patients, mais l’identification de l’impact des variants identifiés : sont-ils pathogènes, probablement pathogènes, bénins ou probablement bénins ? De fait, parmi ces 8 000 variants de RYR1 identifiés à ce jour, pour plus de 3 000 d’entre eux, on ne sait pas s’ils sont, surement ou probablement, pathogènes ou bénins. Ils sont donc classés comme « variants de signification inconnue » (VSI) (Figure 2) et 90 % des variants faux-sens sont de signification inconnue.
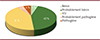 |
Figure 2 Classification actuelle des variants du gène RYR1. VSI : variants de signification inconnue. Graphique réalisé sur la base des variants actuels, recensés dans la base de données ClinVar. |
Le challenge dans la classification des variants génétiques – c’està-dire la capacité à déterminer s’ils sont pathogènes ou non – repose sur plusieurs critères : la complexité des mécanismes pathologiques impliquant parfois plusieurs variants et plusieurs gènes, la variabilité phénotypique entre des patients qui, atteints de la même maladie génétique, peuvent présenter des symptômes différents, et la limitation des outils d’analyse informatique dans leur capacité à prédire avec précision les conséquences des variations [4]. En raison de ces défis, il est souvent nécessaire de mener des études fonctionnelles approfondies pour classer avec précision les VSI et améliorer ainsi le diagnostic des patients.
Objectif du projet
Notre projet de recherche a consisté à élaborer une méthode pour prédire de manière rapide et robuste si tel ou tel variant est potentiellement pathogène ou, au contraire, bénin. Nous avons pour cela réuni les compétences scientifiques de différents domaines : l’intelligence artificielle, la biologie structurale, la physiologie cellulaire et les connaissances cliniques des maladies neuromusculaires. L’objectif : « reclassifier » les VSI du gène RYR1 et classer les variants qui pourraient être nouvellement identifiés.
Méthodologie et résultats préliminaires
Nous avons tout d’abord établi une base de données d’entrainement contenant des variants dont la conséquence a déjà été déterminée précédemment. Cette base compile uniquement des variations « bénignes » et « pathogènes » identifiées par le laboratoire de génétique du Centre hospitalier universitaire de Grenoble, au cours des vingt dernières années. Ces données ont été obtenues lors de séquençages génétiques de patients atteints de myopathies liées à RYR1, d’hyperthermie maligne et d’hyperthermie d’effort, et validées par des études familiales et cliniques. La base de données a été complétée grâce à des recherches bibliographiques décrivant des variations déjà classées à ce jour [5–7] et par des variants issus d’autres bases de données (Leiden open variation database, Human gene mutation database, GnomadV4).
Nous avons ensuite modélisé l’impact des variations sur la structure et la fonction de la protéine. La structure macromoléculaire est divisée en trois régions : le pore du canal, la région d’activation où les ligands se lient, et une région cytoplasmique impliquée dans la fixation de protéines régulatrices. En raison de sa grande taille, peu d’études ont été menées pour étudier la relation structuredynamique-pathogénicité de RyR1. Plus de 90 structures de cette protéine ont été identifiées par des techniques de cryo-microscopie électronique ou de diffraction par rayons X, les plus complètes ayant été décrites pour la protéine RyR1 du lapin [8, 9]. En revanche, en raison de la taille importante de la protéine et de sa flexibilité interne, la structure de nombreuses boucles cytosoliques n’a pas pu être déterminée pour la forme humaine de RyR1. En se basant sur les dernières données expérimentales disponibles dans la littérature, nous avons donc utilisé plusieurs algorithmes pour modéliser la structure de la protéine humaine.
Pour créer les catégories utilisées dans la classification, nous avons ensuite eu besoin de définir différents paramètres appelés « descripteurs ». Un descripteur est un paramètre quantifiable, utilisé pour décrire ou représenter un phénomène de façon simplifiée. Cela facilite son étude par des modèles mathématiques ou informatiques. Dans le contexte de notre étude, les descripteurs sont, par exemple, la conservation, la position de la variation dans la séquence (distance aux différents ligands et domaines fonctionnels), l’impact biochimique du changement d’acide aminé, la modification de la stabilité énergétique de la protéine, la déstabilisation de la protéine, la modification de l’accessibilité du résidu. Le tout, soit pour la forme fermée du canal, soit pour sa forme ouverte.
Grâce à ces descripteurs, chacun des variants de notre base de données se trouve ainsi caractérisé. Après entrainement, ce jeu de données a permis une première validation du modèle informatique. Ce modèle a ensuite été utilisé sur une base de données plus large contenant des VSI. Chaque VSI a ainsi obtenu un score de prédiction de classification qui a établi s’il est potentiellement pathogène ou bénin. Enfin, le processus de classification par intelligence artificielle a été validé par des expériences de biologie sur cellules, une étape essentielle pour confirmer que les prédictions informatiques sont fidèles à l’effet physiopathologique de la variation du gène RYR1. Pour cela, nous avons étudié l’effet physiologique de plusieurs variants qui avaient obtenu une classification – bénin ou pathogène – avec un haut niveau de confiance, renforçant l’apprentissage de notre modèle informatique et améliorant la classification (processus de machine learning) en utilisant à la fois la prédiction informatique et les données expérimentales.
Ces validations fonctionnelles ont débuté avec la réalisation des clonages moléculaires dans un plasmide codant pour la protéine RyR1, afin d’introduire les différents variants dans la séquence du plasmide. Une fois ce travail effectué, nous avons réalisé des expériences d’imagerie calcique sur des lignées cellulaires qui n’expriment pas la protéine RyR1 de façon endogène. Puis, les cellules ont été transfectées avec les plasmides codant pour les différents variants (Figure 3). L’imagerie calcique permet en effet de mettre en évidence des éventuels défauts du relargage du Ca2+ par RyR1, suite à une stimulation du canal par un agoniste spécifique [10, 11]. Nos résultats montrent que cette méthode permet de confirmer la pathogénicité ou l’innocuité d’une mutation de RYR1 sur la libération du Ca2+. Et bien qu’il ne soit pas possible de vérifier fonctionnellement les effets de chaque variant, nos expériences d’imagerie calcique permettent de renforcer les résultats obtenus par les prédictions informatiques.
 |
Figure 3 Graphique représentant les résultats obtenus suite à une expérience d’imagerie calcique. L’amplitude des courbes permet d’apprécier la quantité de Ca2+ libérée dans le cytoplasme en réponse à une stimulation du canal RyR1 avec un agoniste spécifique, le 4-chloro-m-cresol (4CmC). Ici, sont représentées les courbes de réponse de cellules non transfectées (gris), transfectées avec un plasmide codant pour la forme humaine normale de RyR1 (vert), transfectées avec un plasmide codant pour un variant bénin (bleu) ou pathogène (rouge). |
Conclusion et perspectives
Face à la complexité et au nombre croissant de VSI découverts lors de séquençages génétiques, il est nécessaire de développer des outils de classification innovants pour fournir un diagnostic plus précis et rapide aux patients atteints de pathologies associées au gène RYR1. Ainsi, en combinant l’intelligence artificielle, la biologie structurale et les expertises cliniques, moléculaires et fondamentales en maladies neuromusculaires, nous avons développé un modèle de classification robuste pour prédire la pathogénicité ou non des VSI.
Enfin, notre projet contribue à faire le lien entre la génétique et la clinique et notre approche méthodologique pourrait être étendue à d’autres gènes et maladies génétiques, ouvrant ainsi de nouvelles perspectives pour le diagnostic des maladies rares.
Prix SFM
Robin Reynaud Dulaurier a reçu le prix du Meilleur poster lors des journées de la Société française de myologie (SFM) 2022.
Remerciements
Cette étude a reçu le soutien du Programme prioritaire de recherche maladies rares du Programme français d’investissements d’avenir et de l’AFM-Téléthon.
Liens d’intérêt
Les auteur(e)(s) déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Beaufils M, Travard L, Rendu J, et al. Therapies for RYR1-Related Myopathies: Where We Stand and the Perspectives. Curr Pharm Des 2022 ; 28 (1) : 1525. [CrossRef] [PubMed] [Google Scholar]
- « ClinVar ». Consulté le 29 avril 2024. En ligne]. https://www.ncbi.nlm.nih.gov/clinvar/ [Google Scholar]
- Lawal TA, Todd JJ, Meilleur KG. Ryanodine Receptor 1-Related Myopathies: Diagnostic and Therapeutic Approaches. Neurotherapeutics 2018 ; 15 (4) : 885899. [CrossRef] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015 ; 17 (5) : 405–24. [CrossRef] [PubMed] [Google Scholar]
- Kushnir A, Todd JJ, Witherspoon JW, et al. Intracellular calcium leak as a therapeutic target for RYR1-related myopathies. Acta Neuropathol 2020 ; 139 (6) : 1089–1104. [CrossRef] [PubMed] [Google Scholar]
- Garibaldi M, Rendu J, Brocard J, et al. ‘Dusty core disease’ (DuCD): expanding morphological spectrum of RYR1 recessive myopathies. Acta Neuropathol Commun 2019 ; 7 (1) : 3. [CrossRef] [PubMed] [Google Scholar]
- Abath Neto O, de Araújo Martins Moreno C, Malfatti E, et al. Common and variable clinical, histological, and imaging findings of recessive RYR1-related centronuclear myopathy patients. Neuromuscul Disord 2017 ; 27 (11) : 975985. [CrossRef] [PubMed] [Google Scholar]
- Meissner G. The structural basis of ryanodine receptor ion channel function. J Gen Physiol 2017 ; 149 (12) : 10651089. [CrossRef] [PubMed] [Google Scholar]
- Lanner JT, Georgiou DK, Joshi AD, et al. Ryanodine Receptors: Structure, Expression, Molecular Details, and Function in Calcium Release. Cold Spring Harb Perspect Biol 2010 ; 2 (11) : a003996. [CrossRef] [PubMed] [Google Scholar]
- Rendu J, Brocard J, Denarier E, et al. Exon Skipping as a Therapeutic Strategy Applied to an RYR1 Mutation with Pseudo-Exon Inclusion Causing a Severe Core Myopathy. Hum Gene Ther 2013 ; 24 (7) : 702713. [CrossRef] [PubMed] [Google Scholar]
- Noda Y, Miyoshi H, Benucci S, et al. Functional characterization of RYR1 variants identified in malignant hyperthermia susceptible individuals. Neuromuscul Disord 2023 ; 33 (12) : 951963. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1 Le complexe de relargage calcique (CRC) des cellules musculaires. Représentation schématique du complexe de relargage calcique (CRC) à la triade. La protéine RyR1 est ancrée dans la membrane du réticulum sarcoplasmique (RS) et libère le calcium (Ca2+) dans le cytoplasme, en réponse à un potentiel d’action produit à la terminaison axonale. L’augmentation du Ca2+ intracellulaire induit le glissement de la myosine sur les filaments d’actine et la contraction musculaire. Des variations sur le gène RYR1 peuvent conduire à un défaut de libération du Ca2+ et à une perturbation du couplage excitation/contraction. |
| Dans le texte | |
|
Figure 2 Classification actuelle des variants du gène RYR1. VSI : variants de signification inconnue. Graphique réalisé sur la base des variants actuels, recensés dans la base de données ClinVar. |
| Dans le texte | |
|
Figure 3 Graphique représentant les résultats obtenus suite à une expérience d’imagerie calcique. L’amplitude des courbes permet d’apprécier la quantité de Ca2+ libérée dans le cytoplasme en réponse à une stimulation du canal RyR1 avec un agoniste spécifique, le 4-chloro-m-cresol (4CmC). Ici, sont représentées les courbes de réponse de cellules non transfectées (gris), transfectées avec un plasmide codant pour la forme humaine normale de RyR1 (vert), transfectées avec un plasmide codant pour un variant bénin (bleu) ou pathogène (rouge). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.