")
")
| Issue |
Med Sci (Paris)
Volume 40, Number 1, Janvier 2024
La cavité orale et les dents au cœur de la santé
|
|
|---|---|---|
| Page(s) | 64 - 71 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2023197 | |
| Published online | 01 February 2024 | |
Spécificités de la douleur neuropathique oro-faciale
Specificities of orofacial neuropathic pain
1
Laboratoire de neurobiologie orofaciale, EA 7543, Université Paris Cité, Paris, France
2
Hôpital Bretonneau, Service de médecine bucco-dentaire, AP-HP, Paris, France
3
Université Clermont Auvergne, CHU Clermont-Ferrand, Inserm, Neuro-Dol, Clermont-Ferrand, France
4
Hôpital Pitié-Salpêtrière, Service de médecine bucco-dentaire, AP-HP, Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les douleurs de la région céphalique – et notamment les douleurs oro-faciales – diffèrent des douleurs spinales sur les plans physiopathologique, clinique, thérapeutique et pronostique. Leur prévalence élevée, leur fort retentissement sur la qualité de vie individuelle et leur impact économique et sociétal important justifient une étude spécifique. Parmi ces douleurs, les douleurs neuropathiques, résultant d’une maladie ou d’un traumatisme du système nerveux trigéminal, sont parmi les plus difficiles à diagnostiquer et à soigner. L’étude des mécanismes neurobiologiques, périphériques et centraux les sous-tendant a permis de nombreuses avancées conceptuelles, cliniques et thérapeutiques, avec, par exemple, la mise en évidence du rôle des cellules nerveuses et non nerveuses, telles que la glie, les immunocytes, les cellules endothéliales vasculaires ou le rôle de la reconfiguration de la circuiterie nerveuse au niveau du complexe sensitif trigéminal, dans la genèse des douleurs neuropathiques post-lésionnelles. Les interactions cellulaires au sein du ganglion trigéminal, susceptibles d’éclairer la compréhension de certaines comorbidités douloureuses dentaires, oculaires ou céphalalgiques, sont également décrites.
Abstract
Head pain and notably orofacial pain differs from spinal pain on pathophysiological, clinical, therapeutic and prognostic levels. Its high prevalence, important impact on quality of life and significant socio-economical burden justify specific study of such type of pain. Among them, neuropathic orofacial pain resulting from disease or trauma of the trigeminal nervous system is among the most difficult types of pain to diagnose and to treat. Deciphering of underlying peripheral and central mechanisms has allowed numerous conceptual, clinical and therapeutic advances, notably the role of neural and non neural cell types, such as glia, immunocytes, vascular endothelial cells or the role of trigeminal sensory complex neural circuitry reconfiguration in the development of post-traumatic trigeminal neuropathic pain. Cellular interactions within the trigeminal ganglion, allowing a better understanding of several painful dental, ocular or cephalalgic comorbidities, are also described.
Ces auteurs ont contribué de façon égale à ce travail
© 2024 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (© Thibault Canceill).
Les douleurs de la région céphalique, notamment les douleurs oro-faciales (DOF), diffèrent des douleurs spinales du fait des spécificités anatomiques et physiologiques du système nociceptif trigéminal [1]. Elles affectent au moins un adulte sur dix [2] et constituent un véritable enjeu de santé publique aux conséquences considérables, tant en termes de qualité de vie individuelle que de répercussions économiques et sociales. On estime que 22 % de la population générale est confrontée à une DOF au moins une fois en six mois [3] et que 7 à 11 % souffrent de DOF chroniques [4]. Comme les douleurs spinales, les DOF peuvent avoir des origines inflammatoires, neuropathiques et/ou nociplastiques. Alors que les DOF d’origine inflammatoire, liées à des lésions et/ou des maladies des tissus bucco-dentaires, sont bien connues et que leur prise en charge est relativement aisée, les DOF neuropathiques résultant d’une maladie ou d’un traumatisme du système nerveux trigéminal, sont de diagnostic et de traitement bien plus complexes [1, 5]. L’étude des mécanismes neurobiologiques sous-tendant le développement de ces douleurs trigéminales complexes, portée notamment par des équipes de l’Inserm et universitaires ces trente dernières années, a permis de nombreuses avancées conceptuelles, cliniques et thérapeutiques [6–8] (→).
(→) Voir la Synthèse de R. Dallel et al., m/s n° 5, mai 2003, page 567
La DOF neuropathique peut ainsi être considérée comme un modèle prototypique d’exploration du système trigéminal, dont nous décrirons les composantes et les problématiques scientifiques actuelles.
Approche expérimentale de la douleur neuropathique oro-faciale
Les modèles animaux de douleurs neuropathiques
De nombreux modèles animaux ont été développés afin d’étudier la physiopathologie des différents types de douleurs neuropathiques. Ces modèles peuvent être subdivisés en modèles étiologiques, reproduisant l’étiologie suspectée de la neuropathie, ou en modèles symptomatiques, reproduisant son tableau clinique (la constriction nerveuse mimant l’hyperalgésie et l’allodynie de la neuropathie post-traumatique secondaire à un trauma).
En 1994, Vos et al. ont proposé un modèle fondé sur la constriction chronique du nerf infra-orbitaire (CCI-ION pour chronic constriction injury of the infra-orbital nerve), branche du nerf maxillaire (V2), chez le rat [9]. Dans ce modèle, à la suite de la lésion, les animaux développent des changements comportementaux similaires à ceux observés chez l’homme, notamment une première période – d’une quinzaine de jours – d’épisodes de « grooming » de la face, un comportement de frottement de la région infra-orbitaire témoignant d’une douleur spontanée, puis une deuxième phase d’hypersensibilité à différents stimulus appliqués dans le territoire lésé (mimant l’hyperalgésie et l’allodynie observées chez l’homme) et/ou à distance du territoire lésé (hyperalgésie secondaire). Ce modèle CCI-ION a été proposé comme une amélioration du modèle sciatique (CCI-SN, pour chronic constriction injury of the sciatic nerve) [10], dont les changements comportementaux étaient difficiles à interpréter du fait de la nature mixte, sensitive et motrice, du nerf sciatique. Le modèle trigéminal a fait l’objet de différentes adaptations techniques, notamment une voie d’abord endobuccale [11], en préservant la région péri-oculaire, ce qui améliore l’analyse comportementale. D’autres modèles lésionnels, tels que la section du nerf alvéolaire [12] ou la constriction partielle du nerf mentonnier [13], ont été développés ultérieurement.
Spécificités du système nociceptif trigéminal
L’existence de modèles lésionnels homologues, CCI-SN et CCI-ION, offre l’opportunité d’explorer les similarités et les différences entre les systèmes nociceptifs spinal et trigéminal. La comparaison des altérations moléculaires et cellulaires observées dans le parenchyme nerveux lésé après CCI-SN [14] et CCI-ION [15], chez le rat, a ainsi permis d’objectiver une infiltration macrophagique plus précoce et plus importante après CCI-ION. Cette différence est vraisemblablement due à une plus grande densité de fibres myélinisées (50 % vs. 20 %) dans le système trigéminal [16], plus sensibles aux effets nocifs de la constriction chronique [17]. La dégénérescence nerveuse locale résultant de la lésion favoriserait la prolifération de macrophages endoneuraux phagocytant les débris de myéline, selon un mécanisme dépendant de l’IL(interleukine)-6 [18]. Des travaux précédents avaient déjà montré une production plus importante d’IL-6 après CC-ION qu’après CCI-SN [19].
Ces données viennent compléter les nombreux travaux ayant montré la spécificité du système nociceptif trigéminal sur les plans moléculaire, transcriptomique, électrophysiologique et clinique [19–23]. À titre d’exemple, la prolifération sympathique observée au sein des ganglions de racine dorsale après CCI-SN n’est pas observée dans le ganglion trigéminal et la sympathectomie n’affecte pas le niveau de décharges ectopiques dans les neurones trigéminaux lésés. Ces décharges sont moins fréquentes dans le modèle CCI-ION que dans le modèle CCI-SN, en raison sans doute d’une dégénérescence plus rapide des neurones ganglionnaires trigéminaux favorisée par la proximité des sites lésionnels [24]. À la suite d’une lésion nerveuse, il existe également une régulation génique qui s’avère différente au sein des neurones sensoriels spinaux et des neurones sensoriels trigéminaux, tant d’un point de vue moléculaire que cinétique [23]. Enfin, les réponses thérapeutiques observées chez les patients souffrant de neuropathies trigéminales semblent moindres (environ 11 % de patients sont répondeurs) que dans le cas de neuropathies spinales équivalentes (20 à 40 % de patients sont répondeurs) [25].
Physiopathologie des douleurs neuropathiques trigéminales
Le contexte clinique
Les douleurs neuropathiques trigéminales les plus fréquentes sont secondaires à une lésion nerveuse accidentelle ou iatrogène provoquée par des soins bucco-dentaires ou chirurgicaux. Sur le plan épidémiologique, ces neuropathies trigéminales douloureuses post-traumatiques (NTDPT) seraient observées après 3 à 5 % des dépulpations dentaires (aussi appelées « dévitalisations ») et dans environ 3 % des cas après un traumatisme majeur (comme une fracture faciale) [25].
La physiopathologie de ces NTDPT implique une cascade d’évènements dans le système nerveux périphérique et/ou central (Figure 1), notamment des altérations biochimiques, moléculaires et fonctionnelles des cellules neuronales et des cellules gliales, vraisemblablement sur un terrain génétique prédisposant [25].
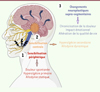 |
Figure 1. Représentation schématique de la chronologie des altérations périphériques puis centrales au sein du système nociceptif trigéminal et leurs répercussions cliniques. |
Sensibilisation périphérique
Une lésion nerveuse entraîne des changements histologiques et électrophysiologiques responsables d’une symptomatologie spécifique (Tableau I). L’hyperactivation des nocicepteurs périphériques, secondaire à l’inflammation locale, conduit à une diminution du seuil d’excitabilité des fibres nerveuses lésées (sensibilisation périphérique). Elle se traduit cliniquement par une hyperalgésie et une allodynie. La régénération des fibres nerveuses lésées peut, par ailleurs, s’accompagner d’une repousse désorganisée et anormale des fibres nerveuses, aboutissant à la formation d’un névrome, de dysesthésies et de paresthésies. La persistance de cette hyperactivité neuronale périphérique, spontanée et provoquée, peut mener à terme à des changements neuroplastiques centraux participant à l’aggravation du tableau clinique et, notamment, à la chronicisation de la douleur [26, 27].
Définition des termes fréquemment employés pour décrire la symptomatologie des douleurs neuropathiques.
Lors d’une lésion nerveuse périphérique, de multiples altérations cellulaires et moléculaires se produisent au sein du parenchyme nerveux, afin de promouvoir la régénération physiologique des fibres nerveuses. Ces altérations peuvent cependant participer également au développement d’une cicatrisation dysfonctionnelle se traduisant, sur le plan phénotypique, par l’apparition d’une douleur neuropathique chronique. La régulation de la perméabilité microvasculaire, au sein de l’endonèvre du nerf lésé, joue un rôle essentiel dans ces phénomènes de cicatrisations nerveuses physiologique et pathologique, via notamment l’infiltration locale de cellules immunitaires réparatrices, successivement des polynucléaires neutrophiles, des macrophages et enfin des lymphocytes T [28].
Le rôle de la disruption précoce de la barrière hémato-nerveuse dans le développement de la neuroinflammation locale post-traumatique a été récemment révélé dans des modèles murins de CCI-SN et de CCI-ION [14, 15]. Cette barrière, équivalent périphérique de la barrière hémato-encéphalique, composée d’une monocouche de cellules endothéliales unies par des protéines de jonctions serrées au sein d’une même membrane basale, permet, en condition physiologique, de protéger le parenchyme nerveux des agressions extérieures, notamment hématogènes. Les voies de signalisation morphogénétiques, Sonic Hedgehog (SHH) et Wnt/β-caténine, ainsi que la voie de l’immunité innée impliquant le récepteur TLR4 (Toll-like receptor 4), participent à la régulation homéostasique de cette barrière au sein des cellules endothéliales endoneurales. L’altération de ces voies favorise la disruption de la barrière hémato-nerveuse en diminuant la synthèse des protéines de jonctions serrées (occludine, claudines) [15, 29]. Devenue hyperperméable, la barrière permet alors l’infiltration locale de substances algogènes (comme le fibrinogène) et d’immunocytes, notamment de macrophages (Figure 2), à l’origine d’altérations de l’excitabilité des fibres nociceptives périphériques conduisant à l’apparition d’un phénotype douloureux neuropathique (hyperalgésie, allodynie).
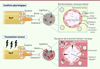 |
Figure 2. Représentation schématique du rôle de la barrière hémato-nerveuse dans le développement de la neuroinflammation locale et l’apparition d’une sensibilisation périphérique. Après traumatisme nerveux, la barrière hémato-nerveuse devient hyperperméable (du fait de la disparition des protéines de jonctions serrées) aux substances algogènes et aux immunocytes. L’infiltration parenchymateuse induit une neuroinflammation locale et l’abaissement du seuil d’excitabilité des fibres nerveuses lésées (sensibilisation périphérique). |
La voie de signalisation morphogénétique SHH participe à de nombreuses étapes de la cicatrisation nerveuse physiologique [30], mais également pathologique comme, notamment la formation de névromes, du fait d’un guidage déficient lors de la repousse axonale [31]. L’exploration de telles voies morphogénétiques (SHH, Wnt/β-caténine, etc.) est ainsi un axe de recherche prometteur afin de comprendre le développement des douleurs neuropathiques post-traumatiques.
Le rôle de la neuroinflammation
Considérant le rôle essentiel de la neuroinflammation locale dans le développement de la douleur neuropathique post-traumatique et notamment l’implication de la voie de signalisation empruntant le récepteur TLR4, la question du rôle initiateur, ou promoteur, d’une inflammation locale préexistante dans les altérations neuropathiques post-traumatiques a été posée. Il a été montré, dans un modèle préclinique de « priming inflammatoire », par injections périneurales répétées de lipopolysaccharide (LPS, un agoniste bactérien du TLR4), que l’existence d’un contexte inflammatoire, avant la survenue du traumatisme nerveux, favorise l’apparition d’un phénotype neuropathique plus précoce et plus sévère après CCI-ION [32]. Cela rejoint le constat clinique fréquent de la prédominance des NTDPT sur des terrains douloureux et/ou inflammatoires préexistants, tels que des infections dentaires ou parodontales [33].
La sensibilisation centrale
La sensibilisation centrale se retrouve dans tous les types de douleurs, qu’elles soient par excès de nociception (après lésion tissulaire ou au cours de l’inflammation), neuropathiques ou cancéreuses, ou même sans lésion organique, comme dans le cas de la migraine ou de la fibromyalgie. Sur le plan clinique, la sensibilisation centrale s’accompagne de l’apparition de divers symptômes douloureux : douleur spontanée, hyperalgésie secondaire, allodynie mécanique ou thermique, douleur référée, douleur diffuse, douleur généralisée. Sur le plan électrophysiologique, cette sensibilisation se traduit principalement par une augmentation de l’activité spontanée et des réponses évoquées des neurones nociceptifs du système nerveux central segmentaire, mais aussi suprasegmentaire, ainsi que par une extension du champ de leurs récepteurs. Bien que la sensibilisation centrale soit à l’origine de la majorité des douleurs chroniques, de nombreux mécanismes physiopathologiques la sous-tendent : augmentation de la transmission excitatrice glutamatergique dépendant du NMDA (acide N-méthyl-D-aspartique), altération des contrôles inhibiteurs et/ou des altérations des interactions neuro-gliales [34].
Au niveau du système nerveux central segmentaire, les fibres afférentes primaires projettent sur le sous-noyau caudal trigéminal qui est organisé en couches cytoarchitectoniques distinctes sur les plans anatomique, électrophysiologique ou fonctionnel (Figure 3). Alors que les fibres Aβ, répondant au tact léger, projettent dans les couches profondes du sous-noyau caudal (III, IV et V), les fibres nociceptives Aδ et C projettent dans les couches I et II externe principalement. À l’interface entre couches superficielles et couches profondes, se trouvent des interneurones excitateurs (exprimant l’isoforme γ de la protéine kinase C [PKCγ]) dans la couche II interne. Ces interneurones sont contactés par des fibres A tactiles et projettent vers les couches superficielles, mais ils reçoivent une inhibition tonique GABAergique et glycinergique empêchant ainsi l’activation des neurones nociceptifs par le toucher [35, 36]. En condition pathologique, notamment lésionnelle, la perte du tonus inhibiteur exercé sur les interneurones PKCγ permet ainsi aux mécanorécepteurs à bas seuil d’accéder au système nociceptif des couches superficielles, entraînant ainsi une allodynie mécanique [35–38]. Ces interneurones PKCγ sont également sous contrôle descendant sérotoninergique (via les récepteurs 5-HT2A) dont l’altération, en contexte pathologique, aboutit à leur activation et à l’apparition d’une allodynie [39]. Cette sensibilisation centrale hétérosynaptique, induite notamment par la libération de dérivés réactifs de l’oxygène suite à l’activation des nocicepteurs, se manifeste quelques secondes après l’application du stimulus conditionnant, mais peut durer plusieurs heures [36]. Si le stimulus est maintenu, même à bas bruit, la sensibilisation centrale persistera d’autant plus. Ainsi, après lésion nerveuse périphérique, l’activité ectopique des nocicepteurs peut conduire à une sensibilisation centrale prolongée.
 |
Figure 3. Représentation schématique et simplifiée des principaux mécanismes à l’origine de la sensibilisation centrale au niveau segmentaire. Celle-ci se manifeste par une désinhibition GABA/glycinergique au niveau segmentaire (représenté en rouge), notamment par la libération de dérivés réactifs de l’oxygène (ROS) par les afférences nociceptives. Les fibres tactiles activent ainsi les interneurones exprimant l’isoforme gamma de la protéine kinase C (PKCγ), notamment au travers de la voie de signalisation NMDA (représentée en vert). Les interactions neuro-gliales (représentées en violet), favorisent l’hyperexcitabilité neuronale, notamment par la libération de D-sérine au niveau synaptique. L’altération des contrôles descendants, notamment sérotoninergiques (représentés en gris), participe également au maintien de la sensibilisation centrale. |
Le principal neurotransmetteur excitateur du système nerveux central, le glutamate, assure la transmission synaptique entre les terminaisons des nocicepteurs et les neurones nociceptifs du sous-noyau caudal. Le glutamate est également libéré par les fibres non nociceptives et les interneurones excitateurs. Sa libération évoque des potentiels post-synaptiques excitateurs rapides dans les neurones des cornes dorsales, en activant des récepteurs de types AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) et kaïnate. En situation normale, le récepteur du glutamate de type NMDA, bien que présent à la synapse, contribue peu aux réponses nociceptives car son canal est bloqué de façon tonique par du magnésium (Mg2+) extracellulaire. En revanche, une neuropathie post-traumatique peut induire une hyperexcitabilité des neurones du sous-noyau caudal dépendante des récepteurs NMDA, notamment suite à une diminution des contrôles inhibiteurs segmentaires et une libération du bouchon de Mg2+ [35, 40].
On a longtemps pensé que les cellules gliales n’avaient qu’un rôle de soutien des neurones, d’intendance et de clairance. On sait maintenant qu’elles sont également des partenaires actifs de la transmission synaptique trigéminale, grâce à leurs récepteurs membranaires et du fait de la libération de gliotransmetteurs qui modulent, en retour, la transmission synaptique. L’altération des interactions entre cellules neuronales et cellules gliales participe à l’apparition de la sensibilisation centrale, via l’implication de la microglie et des astrocytes. En condition physiologique, les cellules microgliales fonctionnent comme des macrophages résidents du système nerveux central, jouant un rôle de sentinelle en cas d’infection ou de lésion. Dans les heures suivant une lésion nerveuse périphérique, la microglie prolifère dans la corne dorsale, en regard des terminaisons des fibres nerveuses lésées et s’active en réponse à la détection de facteurs libérés à la suite de la lésion tissulaire, notamment l’ATP, qui active les récepteurs purinergiques (P2X4, P2X7 et P2Y12 principalement) ou la chimiokine CCL2 (C-C motif chemokine ligand 2) qui se lie au récepteur CXCR2 (C-X-C motif chemokine receptor 2) [41]. La microglie ainsi activée libère de nombreuses cytokines pro-inflammatoires (TNF-α [tumor necrosis factor alpha], IL-1β, IL-6), sensibilisant les neurones nociceptifs centraux, ce qui amplifie le message douloureux. L’activation de P2X4 par l’ATP induit en outre une libération de BDNF (brain-derived neurotrophic factor) par la microglie, dont la fixation à son récepteur post-synaptique TrkB (tropomyosin kinase receptor B) est à l’origine d’une modification du gradient de chlore (Cl-), menant à un changement de l’effet du GABA qui, dans certains cas, peut même devenir dépolarisant (et non plus hyperpolarisant). Ainsi, un stimulus présynaptique GABAergique normalement inhibiteur se traduira par une activation paradoxale du neurone post-synaptique.
L’activation astrocytaire est plus tardive que l’activation microgliale mais elle persiste bien plus longtemps (jusqu’à plusieurs mois), jouant un rôle plutôt dans le maintien de la sensibilisation centrale et la douleur persistante que dans son initiation [42–44]. Les astrocytes jouent également un rôle prépondérant dans l’apparition de douleurs à distance du site de lésion (zone d’hyperalgésie secondaire). En effet, l’activation des astrocytes, dont l’activité syncytiale régule l’homéostasie extracellulaire, notamment à travers la recapture d’ions et de neurotransmetteurs [45], peut influencer l’excitabilité de neurones localisés non seulement dans le territoire correspondant au site lésé, mais également dans des régions adjacentes ou du côté controlatéral au trauma nerveux. Enfin, après une lésion nerveuse périphérique, une activation microgliale et astrocytaire se produit également dans différentes structures suprasegmentaires, notamment dans la substance grise périaqueducale, liées à la transmission ou à la modulation des messages nociceptifs, conduisant également à une hyperexcitabilité des voies nociceptives trigéminales [46, 47] (Figure 3).
Le rôle du ganglion trigéminal dans la physiopathologie des douleurs neuropathiques trigéminales
Le ganglion trigéminal est le ganglion sensitif contenant les corps cellulaires des neurones des trois branches trigéminales : ophtalmique (V1), maxillaire (V2) et mandibulaire (V3). La branche V1 assure l’innervation des méninges, du front, de la région orbitaire, de la cornée, de la région temporale supérieure et antérieure, de la racine du nez et de la muqueuse nasale. La branche V2 innerve les téguments de la lèvre supérieure, de la joue, de la paupière inférieure, de la région temporale, des gencives, de la cloison des fosses nasales, du palais et des 2/3 antérieurs du rhino-pharynx et de la muqueuse du sinus maxillaire. Enfin la branche V3 innerve les téguments de la tempe, du menton, des dents et gencives de la mâchoire inférieure, des 2/3 antérieurs de la langue et de la muqueuse buccale et des joues. Les neurones du ganglion trigéminal ont une structure pseudo-unipolaire, avec une extrémité projetant vers la périphérie et l’autre vers le complexe sensitif du trijumeau localisé au sein du tronc cérébral. Le ganglion trigéminal est ainsi schématiquement au centre du système trigéminal, à l’interface entre le système nerveux périphérique et le système nerveux central. Bien que son implication dans les douleurs neuropathiques oro-faciales soit connue depuis plus d’un siècle, son rôle précis dans leur physiopathologie commence seulement à être élucidé.
La ganglionectomie trigéminale : un traitement historique de la névralgie trigéminale
La névralgie trigéminale est la première douleur neuropathique trigéminale qui a été décrite de manière détaillée, en 1671, mais elle est connue depuis l’antiquité [48]. C’est une maladie neurologique complexe affectant le système trigéminal, caractérisée par des crises douloureuses paroxystiques de courte durée, sans douleur entre les crises, pouvant affecter une ou plusieurs branches du nerf trijumeau. Sa physiopathologie est encore imparfaitement élucidée mais elle impliquerait des décharges électriques éphaptiques liées à une démyélinisation focale secondaire à une compression vasculaire sur le trajet du nerf trijumeau dans sa portion intracrânienne [49]. Certaines formes cliniques non liées à un conflit neuro-vasculaire impliqueraient des mutations de certains canaux ioniques (canaux sodiques voltage dépendants, canaux calciques, canaux chlore, canaux TRP [transient receptor potential]) se traduisant par une hyperexcitabilité anormale des neurones trigéminaux et expliquant la présence de formes familiales de névralgie trigéminale [50].
Un des premiers traitements de la névralgie trigéminale était la ganglionectomie trigéminale, décrite par le neurochirurgien Harvey Cushing dès 1900. Son efficacité, notamment en cas d’échec d’une neurotomie périphérique d’une ou plusieurs branches trigéminales, illustre le rôle central de cette structure dans la physiopathologie de la névralgie trigéminale.
Le rôle de la neuroinflammation et des interactions neuro-gliales ganglionnaires
Le ganglion trigéminal contient les corps cellulaires des neurones, tous entourés de cellules satellites gliales (CSG). Des études récentes utilisant des modèles murins ex vivo (cocultures de cellules trigéminales) et in vivo (stimulation trigéminale douloureuse) suggèrent une possible activation croisée entre les différentes branches nerveuses au sein du ganglion trigéminal. Celle-ci résulterait d’interactions neuro-gliales entre les corps cellulaires des neurones trigéminaux et les CSG les entourant, via des neuropeptides tels que le CGRP (calcitonin gene-related peptide), la substance P et le PACAP (pituitary adenylate cyclase-activating polypeptide), ainsi que d’autres molécules de signalisation (ATP, monoxyde d’azote [NO], cytokines, neurotrophines, etc.) [51]. À la suite d’une lésion nerveuse, les CSG sont activées (avec propagation de l’activation aux CSG voisines par surexpression de la protéine de jonction Cx-43 [connexin 43] [52]), ce qui se traduit par une augmentation de la production de certains gliomarqueurs, tels que la GFAP (glial fibrillary acidic protein) [53] et la synthèse de chimiokines et de cytokines pro-inflammatoires, telles que CCL2 et l’IL-1β [41]. Cette activation apparaît très rapidement (quatre heures post-lésion) avec un maximum à une semaine, puis diminue après trois semaines. Cette cinétique d’activation suggère un rôle prépondérant des CSG lors des premières phases du développement de la neuropathie trigéminale post-traumatique [53]. Plus récemment, des auteurs ont montré qu’il était possible d’atténuer les symptômes douloureux provoqués par la lésion, en inhibant, par une approche chémogénétique (ou DREADD, pour designer receptor exclusively activated by designer drugs), les CSG du ganglion trigéminal [54].
Le système nociceptif cornéen
La nature précise des interactions fonctionnelles entre les différentes branches trigéminales n’a que peu été étudiée, en particulier dans le contexte spécifique des NTDPT. Leur étude est d’autant plus importante que le ganglion trigéminal est au centre de la physiopathologie de nombreuses pathologies douloureuses, notamment les douleurs inflammatoires et neuropathiques faciales, mais également les céphalées. Des données préliminaires suggèrent, par ailleurs, que les sensibilisations périphérique et centrale résultant d’une lésion neuropathique de la branche V2, pourrait participer à la sensibilisation de la branche V1 dans un modèle murin de migraine [55]. Afin d’explorer les interactions moléculaires et fonctionnelles entre les différentes branches trigéminales, il a été récemment proposé l’étude du système nociceptif cornéen. En effet, la cornée – tissu le plus innervé de l’organisme – reçoit son innervation des nerfs ciliaires, eux-mêmes issus de la branche V1. Du fait de la transparence de la cornée, cette innervation peut être explorée en temps réel grâce à des techniques ophtalmologiques spécifiques non invasives, telles que la microscopie confocale in vivo, tant chez l’homme que chez l’animal [56]. À ce titre, l’exploration des altérations du système nociceptif cornéen dans des modèles animaux lésionnels comme la CCI-ION, devrait permettre de mieux comprendre les interactions fonctionnelles entre les branches trigéminales et les altérations ganglionnaires impliquées dans ces interactions (Figure 4).
 |
Figure 4. Approche expérimentale pour étudier les interactions fonctionnelles et moléculaires entre les différentes branches trigéminales en condition lésionnelle (ici une neuropathie par constriction chronique du nerf infra-orbitaire). Après avoir induit la lésion neuropathique (1), les répercussions sur le système nociceptif cornéen seront explorées (2), notamment via la microscopie confocale in vivo permettant une imagerie non invasive des structures et composants de la cornée (comme par exemple ici des interactions étroites entre les nerfs cornéens et les kératocytes activés, témoignant d’altérations cornéennes [branche V1] secondaires à la neuropathie [branche V2]). Enfin, les altérations moléculaires intra-ganglionnaires seront étudiées (3) pour comprendre les mécanismes expliquant l’interaction V2/V1 observée. |
Des données préliminaires ont permis d’objectiver des altérations cornéennes ipsilatérales post CCI-ION, notamment des ulcérations cornéennes et une hypoesthésie cornéenne mécanique. Ces altérations pourraient être dues à la perte du soutien trophique des neurones cornéens, dont le corps cellulaire a été altéré indirectement par la lésion de la branche V2 (données non publiées). Les mécanismes précis sous-tendant l’interaction V2/V1 restent à explorer. Cette approche expérimentale pourrait, entre autres, ouvrir la voie à l’exploration de complications ophtalmologiques des NTDPT, encore méconnues.
Conclusion et perspectives
Le système trigéminal possède des spécificités anatomiques et physiologiques ainsi que des spécificités cliniques, avec des douleurs chroniques sous-tendues par des mécanismes physiopathologiques propres nécessitant des thérapeutiques particulières. Les travaux récents ont amélioré notre compréhension de la physiopathologie des douleurs trigéminales et ont notamment permis d’identifier certaines particularités du système nociceptif trigéminal. La création de modèles murins spécifiques et les innovations technologiques récentes devraient aider à mieux caractériser les processus de chronicisation des douleurs trigéminales, leur forte association avec des troubles psychiatriques ainsi que leur forte prévalence féminine.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Moreau N, Boucher Y. Douleurs orofaciales. Encyclopédie Médico Chirurgicale 2020; 28–290-C-10. [Google Scholar]
- Kohlmann T. Epidemiology of orofacial pain. Schmerz 2002 ; 16 : 339–345. [CrossRef] [PubMed] [Google Scholar]
- Lipton JA, Ship JA, Larach-Robinson D. Estimated prevalence and distribution of reported orofacial pain in the United States. J Am Dent Assoc 1993 ; 124 : 115–121. [CrossRef] [PubMed] [Google Scholar]
- Goulet J-P, Woda A. Orofacial Pain: Classification and Road Map to Clinical Phenotypes. In : Goulet J-P, Velly AM, editors. Orofacial Pain Biomarkers. Berlin, Heidelberg: Springer, 2017 : 3–20. [CrossRef] [Google Scholar]
- Moreau N, Boucher Y. A-t-on perdu la tête ? Plaidoyer pour l’étude et la prise en charge des douleurs orofaciales. Douleur analg 2022; 35 : 43–5. [CrossRef] [Google Scholar]
- Dallel R, Voisin D. Towards a pain treatment based on the identification of the pain-generating mechanisms?. Eur Neurol 2001 ; 45 : 126–132. [CrossRef] [PubMed] [Google Scholar]
- Dallel R, Villanueva L, Woda A, et al. Neurobiologie de la douleur trigéminale. Med Sci (Paris) 2003 ; 19 : 567–574. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Moreau N, Dieb W, Descroix V, et al. Topical Review: Potential Use of Botulinum Toxin in the Management of Painful Posttraumatic Trigeminal Neuropathy. J Oral Facial Pain Headache 2017 ; 31 : 7–18. [CrossRef] [PubMed] [Google Scholar]
- Vos BP, Strassman AM, Maciewicz RJ, Behavioral evidence of trigeminal neuropathic pain following chronic constriction injury to the rat’s infraorbital nerve. J Neurosci 1994 ; 14 : 2708–2723. [CrossRef] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK, A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988 ; 33 : 87–107. [CrossRef] [PubMed] [Google Scholar]
- Imamura Y, Kawamoto H, Nakanishi O, Characterization of heat-hyperalgesia in an experimental trigeminal neuropathy in rats. Exp Brain Res 1997 ; 116 : 97–103. [CrossRef] [PubMed] [Google Scholar]
- Iwata K, Imai T, Tsuboi Y, et al. Alteration of medullary dorsal horn neuronal activity following inferior alveolar nerve transection in rats. J Neurophysiol 2001 ; 86 : 2868–2877. [CrossRef] [PubMed] [Google Scholar]
- Boucher Y, Carstens MI, Sawyer CM, et al. Capsaicin avoidance as a measure of chemical hyperalgesia in orofacial nerve injury models. Neurosci Lett 2013 ; 543 : 37–41. [CrossRef] [PubMed] [Google Scholar]
- Moreau N, Mauborgne A, Bourgoin S, et al. Early alterations of Hedgehog signaling pathway in vascular endothelial cells after peripheral nerve injury elicit blood-nerve barrier disruption, nerve inflammation, and neuropathic pain development. Pain 2016 ; 157 : 827–839. [CrossRef] [PubMed] [Google Scholar]
- Moreau N, Dieb W, Mauborgne A, et al. Hedgehog Pathway-Mediated Vascular Alterations Following Trigeminal Nerve Injury. J Dent Res 2017 ; 96 : 450–457. [CrossRef] [PubMed] [Google Scholar]
- Jacquin MF, Hess A, Yang G, et al. Organization of the infraorbital nerve in rat: a quantitative electron microscopic study. Brain Res 1984 ; 290 : 131–135. [CrossRef] [PubMed] [Google Scholar]
- Guilbaud G, Gautron M, Jazat F, et al. Time course of degeneration and regeneration of myelinated nerve fibres following chronic loose ligatures of the rat sciatic nerve: can nerve lesions be linked to the abnormal pain-related behaviours?. Pain 1993 ; 53 : 147–158. [CrossRef] [PubMed] [Google Scholar]
- Ko JS, Eddinger KA, Angert M, et al. Spinal activity of interleukin 6 mediates myelin basic protein-induced allodynia. Brain Behav Immun 2016 ; 56 : 378–389. [CrossRef] [PubMed] [Google Scholar]
- Latrémolière A, Mauborgne A, Masson J, et al. Differential implication of proinflammatory cytokine interleukin-6 in the development of cephalic versus extracephalic neuropathic pain in rats. J Neurosci 2008 ; 28 : 8489–8501. [CrossRef] [PubMed] [Google Scholar]
- Hargreaves KM, Orofacial pain. Pain 2011 ; 152 : S25–S32. [CrossRef] [PubMed] [Google Scholar]
- Benoliel R, Zadik Y, Eliav E, et al. Peripheral painful traumatic trigeminal neuropathy: clinical features in 91 cases and proposal of novel diagnostic criteria. J Orofac Pain 2012 ; 26 : 49–58. [PubMed] [Google Scholar]
- Megat S, Ray PR, Tavares-Ferreira D, et al. Differences between Dorsal Root and Trigeminal Ganglion Nociceptors in Mice Revealed by Translational Profiling. J Neurosci 2019 ; 39 : 6829–6847. [CrossRef] [PubMed] [Google Scholar]
- Korczeniewska OA, Khan J, Eliav E, et al. Molecular mechanisms of painful traumatic trigeminal neuropathy-Evidence from animal research and clinical correlates. J Oral Pathol Med 2020; 49 : 580–9. [CrossRef] [PubMed] [Google Scholar]
- Tal M, Devor M, Ectopic discharge in injured nerves: comparison of trigeminal and somatic afferents. Brain Res 1992 ; 579 : 148–151. [CrossRef] [PubMed] [Google Scholar]
- Baad-Hansen L, Benoliel R, Neuropathic orofacial pain: Facts and fiction. Cephalalgia 2017 ; 37 : 670–679. [CrossRef] [PubMed] [Google Scholar]
- Xie W, Strong JA, Meij JTA, et al. Neuropathic pain: early spontaneous afferent activity is the trigger. Pain 2005 ; 116 : 243–256. [CrossRef] [PubMed] [Google Scholar]
- Baron R, Hans G, Dickenson AH, Peripheral input and its importance for central sensitization. Ann Neurol 2013 ; 74 : 630–636. [CrossRef] [PubMed] [Google Scholar]
- Sapienza ARéaux-Le Goazigo A, Rostène W, et al. [Chemokines and attraction of myeloid cells in peripheral neuropathic pains]. Biol Aujourdhui 2014 ; 208 : 31–44. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Moreau N, Mauborgne A, Couraud P-O, et al. Could an endoneurial endothelial crosstalk between Wnt/β-catenin and Sonic Hedgehog pathways underlie the early disruption of the infra-orbital blood-nerve barrier following chronic constriction injury?. Mol Pain 2017 ; 13 : 1744806917727625. [CrossRef] [Google Scholar]
- Moreau N, Boucher Y. Hedging against Neuropathic Pain: Role of Hedgehog Signaling in Pathological Nerve Healing. Int J Mol Sci 2020; 21 : 9115. [CrossRef] [PubMed] [Google Scholar]
- Yamada Y, Ohazama A, Maeda T, et al. The Sonic Hedgehog signaling pathway regulates inferior alveolar nerve regeneration. Neurosci Lett 2018 ; 671 : 114–119. [CrossRef] [PubMed] [Google Scholar]
- Boucher Y, Moreau N, Mauborgne A, et al. Lipopolysaccharide-mediated inflammatory priming potentiates painful post-traumatic trigeminal neuropathy. Physiol Behav 2018 ; 194 : 497–504. [CrossRef] [PubMed] [Google Scholar]
- Dieb W, Moreau N, Chemla I, et al. Neuropathic pain in the orofacial region: The role of pain history. A retrospective study. J Stomatol Oral Maxillofac Surg 2017 ; 118 : 147–150. [CrossRef] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ, Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain 2009 ; 10 : 895–926. [CrossRef] [PubMed] [Google Scholar]
- Miraucourt LS, Dallel R, Voisin DL, Glycine inhibitory dysfunction turns touch into pain through PKCgamma interneurons. PLoS ONE 2007 ; 2 : e1116. [Google Scholar]
- Peirs C, Bourgois N, Artola A, et al. Protein Kinase C γ Interneurons Mediate C-fiber-induced Orofacial Secondary Static Mechanical Allodynia, but Not C-fiber-induced Nociceptive Behavior. Anesthesiology 2016 ; 124 : 1136–1152. [CrossRef] [PubMed] [Google Scholar]
- Artola A, Voisin D, Dallel R. PKCγ interneurons, a gateway to pathological pain in the dorsal horn. J Neural Transm (Vienna) 2020; 127 : 527–40. [CrossRef] [PubMed] [Google Scholar]
- El Khoueiry C, Alba-Delgado C, Antri M, et al. GABAA and Glycine Receptor-Mediated Inhibitory Synaptic Transmission onto Adult Rat Lamina IIi PKCγ-Interneurons: Pharmacological but Not Anatomical Specialization. Cells 2022; 11 : 1356. [CrossRef] [PubMed] [Google Scholar]
- Alba-Delgado C, Mountadem S, Mermet-Joret N, et al. 5-HT2A Receptor-Induced Morphological Reorganization of PKCγ-Expressing Interneurons Gates Inflammatory Mechanical Allodynia in Rat. J Neurosci 2018 ; 38 : 10489–10504. [CrossRef] [PubMed] [Google Scholar]
- Torsney C, MacDermott AB, Disinhibition opens the gate to pathological pain signaling in superficial neurokinin 1 receptor-expressing neurons in rat spinal cord. J Neurosci 2006 ; 26 : 1833–1843. [CrossRef] [PubMed] [Google Scholar]
- Dauvergne C, Molet J, Reaux-Le Goazigo A, et al. Implication of the chemokine CCL2 in trigeminal nociception and traumatic neuropathic orofacial pain. Eur J Pain 2014 ; 18 : 360–375. [CrossRef] [PubMed] [Google Scholar]
- Molet J, Mauborgne A, Diallo M, et al. Microglial JAK/STAT3 pathway activity directly impacts astrocyte and spinal neuron characteristics. J Neurochem 2016 ; 136 : 133–147. [CrossRef] [PubMed] [Google Scholar]
- Miraucourt LS, Peirs C, Dallel R, et al. Glycine inhibitory dysfunction turns touch into pain through astrocyte-derived D-serine. Pain 2011 ; 152 : 1340–1348. [CrossRef] [PubMed] [Google Scholar]
- Lefèvre Y, Amadio A, Vincent P, et al. Neuropathic pain depends upon D-serine co-activation of spinal NMDA receptors in rats. Neurosci Lett 2015 ; 603 : 42–47. [CrossRef] [PubMed] [Google Scholar]
- Olsen ML, Khakh BS, Skatchkov SN, et al. New Insights on Astrocyte Ion Channels: Critical for Homeostasis and Neuron-Glia Signaling. J Neurosci 2015 ; 35 : 13827–13835. [CrossRef] [PubMed] [Google Scholar]
- Boyer N, Dallel R, Artola A, et al. General trigeminospinal central sensitization and impaired descending pain inhibitory controls contribute to migraine progression. Pain 2014 ; 155 : 1196–1205. [CrossRef] [PubMed] [Google Scholar]
- Wei F, Guo W, Zou S, et al. Supraspinal glial-neuronal interactions contribute to descending pain facilitation. J Neurosci 2008 ; 28 : 10482–10495. [CrossRef] [PubMed] [Google Scholar]
- Eboli P, Stone JL, Aydin S, et al. Historical characterization of trigeminal neuralgia. Neurosurgery 2009; 64 : 1183–6; discussion 1186–7. [CrossRef] [PubMed] [Google Scholar]
- Bendtsen L, Zakrzewska JM, Heinskou TB, et al. Advances in diagnosis, classification, pathophysiology, and management of trigeminal neuralgia. Lancet Neurol 2020; 19 : 784–96. [CrossRef] [PubMed] [Google Scholar]
- Gualdani R, Gailly P, Yuan J-H, et al. A TRPM7 mutation linked to familial trigeminal neuralgia: Omega current and hyperexcitability of trigeminal ganglion neurons. Proc Natl Acad Sci U S A 2022; 119 : e2119630119. [CrossRef] [PubMed] [Google Scholar]
- Messlinger K, Russo AF, Current understanding of trigeminal ganglion structure and function in headache. Cephalalgia 2019 ; 39 : 1661–1674. [CrossRef] [PubMed] [Google Scholar]
- Komiya H, Shimizu K, Ishii K, et al. Connexin 43 expression in satellite glial cells contributes to ectopic tooth-pulp pain. J Oral Sci 2018 ; 60 : 493–499. [CrossRef] [PubMed] [Google Scholar]
- Ji R-R, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE 2004; 2004 : reE14. [PubMed] [Google Scholar]
- Korczeniewska OA, James MH, Eliav T, et al. Chemogenetic inhibition of trigeminal ganglion neurons attenuates behavioural and neural pain responses in a model of trigeminal neuropathic pain. Eur J Pain 2022; 26 : 634–47. [CrossRef] [PubMed] [Google Scholar]
- Toyama M, Kudo C, Mukai C, et al. Trigeminal nervous system sensitization by infraorbital nerve injury enhances responses in a migraine model. Cephalalgia 2017 ; 37 : 1317–1328. [CrossRef] [PubMed] [Google Scholar]
- Réaux-Le Goazigo A, Melik Parsadaniantz S, Baudouin C, et al. Douleur oculaire : du fondamental à la clinique. Douleurs : Évaluation - Diagnostic - Traitement 2022; 23 : 75–85. [CrossRef] [Google Scholar]
Liste des tableaux
Définition des termes fréquemment employés pour décrire la symptomatologie des douleurs neuropathiques.
Liste des figures
|
Figure 1. Représentation schématique de la chronologie des altérations périphériques puis centrales au sein du système nociceptif trigéminal et leurs répercussions cliniques. |
| Dans le texte | |
|
Figure 2. Représentation schématique du rôle de la barrière hémato-nerveuse dans le développement de la neuroinflammation locale et l’apparition d’une sensibilisation périphérique. Après traumatisme nerveux, la barrière hémato-nerveuse devient hyperperméable (du fait de la disparition des protéines de jonctions serrées) aux substances algogènes et aux immunocytes. L’infiltration parenchymateuse induit une neuroinflammation locale et l’abaissement du seuil d’excitabilité des fibres nerveuses lésées (sensibilisation périphérique). |
| Dans le texte | |
|
Figure 3. Représentation schématique et simplifiée des principaux mécanismes à l’origine de la sensibilisation centrale au niveau segmentaire. Celle-ci se manifeste par une désinhibition GABA/glycinergique au niveau segmentaire (représenté en rouge), notamment par la libération de dérivés réactifs de l’oxygène (ROS) par les afférences nociceptives. Les fibres tactiles activent ainsi les interneurones exprimant l’isoforme gamma de la protéine kinase C (PKCγ), notamment au travers de la voie de signalisation NMDA (représentée en vert). Les interactions neuro-gliales (représentées en violet), favorisent l’hyperexcitabilité neuronale, notamment par la libération de D-sérine au niveau synaptique. L’altération des contrôles descendants, notamment sérotoninergiques (représentés en gris), participe également au maintien de la sensibilisation centrale. |
| Dans le texte | |
|
Figure 4. Approche expérimentale pour étudier les interactions fonctionnelles et moléculaires entre les différentes branches trigéminales en condition lésionnelle (ici une neuropathie par constriction chronique du nerf infra-orbitaire). Après avoir induit la lésion neuropathique (1), les répercussions sur le système nociceptif cornéen seront explorées (2), notamment via la microscopie confocale in vivo permettant une imagerie non invasive des structures et composants de la cornée (comme par exemple ici des interactions étroites entre les nerfs cornéens et les kératocytes activés, témoignant d’altérations cornéennes [branche V1] secondaires à la neuropathie [branche V2]). Enfin, les altérations moléculaires intra-ganglionnaires seront étudiées (3) pour comprendre les mécanismes expliquant l’interaction V2/V1 observée. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.