")
")
| Issue |
Med Sci (Paris)
Volume 37, Novembre 2021
Les Cahiers de Myologie
|
|
|---|---|---|
| Page(s) | 40 - 43 | |
| Section | Cas clinique | |
| DOI | https://doi.org/10.1051/medsci/2021191 | |
| Published online | 08 December 2021 | |
Dystrophie musculaire liée à des mutations du gène JAG2
L’importance du diagnostic différentiel
JAG2-related muscular dystrophy: When differential diagnosis matters
1
Centre de référence des maladies neuromusculaires Nord/Est/Île-de-France, service de neuromyologie, APHP, Institut de Myologie, Paris, France
2
Sorbonne Université - Inserm, Centre de Recherche en Myologie, Paris, France
3
Unité de Morphologie Neuromusculaire, Institut de Myologie, APHP, Sorbonne Université, Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Abstract
JAG2 has recently been involved in autosomal recessive forms of muscular dystrophy as illustrated in this clinical vignette. In many ways, this disease can mimick a COL6-related retractile myopathy including at the imaging level.
© 2021 médecine/sciences – Inserm
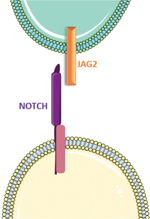
© Valérie Allamand/Servier Medical Art
Observation
Nous présentons le cas d’un patient de 45 ans, premier d’une fratrie de deux enfants issus de parents consanguins d’origine française. Le développement moteur était normal dans les premières années (tenue assise acquise à l’âge de 4 mois, acquisition de la marche à l’âge de 14 mois). Les premiers symptômes sont notés au cours de l’enfance avec des chutes fréquentes, des difficultés pour monter les escaliers ainsi que des difficultés à la course. Le patient a développé progressivement une faiblesse axiale et proximale, une rigidité rachidienne ainsi que des rétractions localisées principalement aux fléchisseurs des doigts et aux tendons d’Achille. La marche a été perdue à l’âge de 14 ans. Il présente également une scoliose nettement accentuée pendant l’adolescence avec nécessité d’une arthrodèse rachidienne à l’âge de 18 ans. Il n’y a pas de difficultés d’apprentissage ni de troubles cognitifs. Une IRM cérébrale se révèle normale. Lors de la dernière consultation à l’âge de 41 ans, l’examen clinique montre une faiblesse motrice à prédominance axiale et proximale des quatre membres. Aux membres supérieurs, le déficit prédomine sur les muscles proximaux cotés à 2/5 au score MRC et une atteinte distale à moindre degré (3-4/5). Aux membres inférieurs, on note un déficit moteur majeur proximo-distal : psoas, fessiers et adducteurs sont à 1/5, le quadriceps à 2/5, les ischiojambiers à 2/5, le jambier antérieur à 1/5 et flexion dorsale à 2/5. Des rétractions articulaires sont observées aux poignets, aux coudes, aux épaules et aux genoux. Il n’y avait pas de lésions cutanées, pas d’atteinte faciale ni troubles de l’oculomotricité. Un syndrome respiratoire restrictif asymptomatique complète ce tableau avec une capacité vitale fonctionnelle évaluée à 55 % de la théorique. Le bilan cardiologique est normal. Les examens paracliniques montrent un taux de CK normal et des tracés myogènes à l’EMG. Une première biopsie musculaire faite à l’âge de 19 ans aurait révélé une fibrose endomysiale ainsi que des fibres en nécrose, l’ensemble ayant fait suspecter initialement une dystrophie musculaire de Duchenne. L’analyse génétique du gène DMD ne montre pas d’anomalies. Les immunomarquages des protéines membranaires sont normaux hormis un discret déficit en a-dystroglycane. Les premières analyses génétiques portent sur les a-dystroglycanopathies (panel comprenant entre autres les gènes FKRP, ISPD, TMEM5, GTDC2, B3GALNT2, SGK196) ainsi que d’autres gènes de pathologies musculaires rétractiles (LMNA, SEPN1 et GAA), et s’avèrent normales. Une deuxième biopsie réalisée à l’âge de 24 ans a montré une inégalité de taille de fibres, une augmentation du tissu conjonctif endomysial mais aussi une certaine désorganisation de la structure myofibrillaire avec présence de fibres lobulées ( Figure 1 ).
 |
Figure 1. Biopsie musculaire (deltoïde). Les colorations hématoxyline-éosine (HE) et trichrome de Gomori (TG) montrent la présence de fibres musculaires de taille inégale avec légère augmentation du tissu conjonctif interstitiel. Certaines fibres montrent un remaniement de la structure et de rares noyaux internalisés. La technique oxydative NADH révèle la présence de fibres musculaires de taille inégale dont nombreuses présentent un aspect lobulé avec désorganisation de la structure. La coloration ATP 9,40 montre des fibres musculaires de taille inégale ainsi que la présence de fibres de petite taille. |
Une IRM musculaire corps entier met en évidence une atrophie et une involution graisseuse très importantes des muscles paravertébraux, ainsi que des muscles fessiers, ceux des loges antérieure et postérieure des cuisses, et à moindre degré des muscles des jambes. On observe une involution graisseuse qui débute à la périphérie des muscles de la cuisse (i.e. vaste externe du quadriceps) et de la jambe (i.e. soléaire, gastrocnémien) et une hypodensité centrale du muscle droit antérieur ( Figure 2 ). Ce tableau clinique évoluant depuis l’enfance, avec atteinte rétractile à prédominance proximale, associé à des images IRM évocatrices, évoque à ce moment précis une myopathie liée à des mutations du collagène VI (COL6-RM). Cependant, l’étude de la sécrétion du collagène VI se révèle normale. L’analyse des gènes COL6A1-A2-A3 ne montre pas non plus de variants pathogènes. Une analyse d’exome entier est entreprise en dernier recours et révèle la présence d’un variant pathogène du gène JAG2 (c.2930T>C, p.Phe977Ser). Il s’agit d’une mutation faux-sens homozygote affectant une région hautement conservée et prédite pathogène par de nombreuses bases de données et algorithmes (PolyPhen2, SIFT, PROVEAN, FATHMM, CADD).
 |
Figure 2. Imagerie musculaire (IRM corps entier). Séquences pondérées en T1. A. Coupe transversale au niveau thoracique montrant une atrophie des muscles paravertébraux ainsi qu’une involution graisseuse très importante au niveau des bras, particulièrement biceps et triceps brachiaux, avec une image d’atrophie « en doigts de gants ». B. Coupe transversale des cuisses montrant une atrophie et une involution graisseuse des deux loges mais qui débute dans la périphérie des muscles (aspect en « sandwich » sur le muscle vaste latéral). On note également le signe de la cible (target sign) sur le droit fémoral. C. Coupe transversale des jambes qui révèle une atrophie et un remplacement graisseux très importants au niveau des muscles gastrocnémiens latéral et médial et à moindre degré des muscles jambiers antérieurs, extenseurs des orteils et péroniers avec relative conservation du muscle tibial postérieur. |
La dystrophie musculaire liée à JAG2
La myopathie liée au gène JAG2 a été décrite très récemment dans une cohorte internationale de vingt-trois patients âgés de 5 à 45 ans, porteurs de mutations récessives dans le gène JAG2, et dont le patient décrit ici faisait partie [1]. JAG2 est situé sur la région chromosomique 14q32.33 et code le ligand de Notch, Jagged2 [2]. Il est exprimé principalement dans le muscle squelettique, le cœur et le pancréas [3]. Les ligands de Notch correspondent à une famille de protéines transmembranaires avec des domaines extracellulaires similaires, tels qu’un domaine N-terminal C2, un domaine DSL (Delta/Serrate/Lag-2) et un nombre variable de répétitions EGF (Epidermal Growth Factor) [4]. Ces ligands interagissent directement avec des récepteurs Notch qui migrent vers le noyau afin d’activer la transcription de gènes cibles. La voie de signalisation Notch est une voie très conservée qui contribue au développement et à l’homéostasie de multiples tissus, dont le muscle squelettique [5-7]. La présence de mutations d’autres ligands de Notch a été associée à des maladies systémiques comme le syndrome d’Alagille (caractérisé par une atteinte hépatique, cardiaque et pulmonaire) ou la tétralogie de Fallot (due à des mutations dominantes de JAG1), à des anomalies du développement du système nerveux central (SNC) (mutations dominantes de DLL1) [8–11], au syndrome CADASIL consécutif aux mutations du gène NOTCH3 (Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) [14], ainsi qu’au syndrome de dysostose spondylo-costale (mutations récessives du gène DLL3) [12]. Par ailleurs, des mutations du gène JAG1 ont été également associées à des neuropathies périphériques héréditaires avec atteinte des cordes vocales [13].
Concernant la dystrophie musculaire liée au gène JAG2, le début des symptômes est variable, s’étendant de la petite enfance à l’adolescence. La faiblesse musculaire prédomine dans la musculature axiale et proximale, notamment aux membres inférieurs. L’évolution est lentement progressive mais une perte de la marche avant l’âge adulte est décrite dans 35 % des cas [1]. Les rétractions sont souvent présentes, affectant principalement les tendons d’Achille et les coudes, mais dans certains cas, le phénotype rétractile peut être plus diffus. La scoliose et la rigidité spinale sont présentes dans la moitié des cas. L’atteinte faciale et le ptosis ont été décrits chez certains patients, mais aucun des patients rapportés jusqu’alors ne présente de troubles de l’oculomotricité. L’atteinte respiratoire est relativement fréquente (65 %), avec une insuffisance respiratoire modérée, souvent asymptomatique, et ne nécessitant pas d’assistance ventilatoire. Une cardiomyopathie ou des troubles du rythme sont également rapportés chez 8 des 23 patients. Il existe également dans certains cas des troubles cognitifs (i.e. difficultés pour l’apprentissage, retard dans l’acquisition du langage, troubles du spectre autistique) sans lésions structurelles du système nerveux central.
Concernant les examens complémentaires, le taux de CPK est habituellement normal ou très faiblement élevé, et l’électromyogramme montre un syndrome myogène. La biopsie musculaire révèle des anomalies myopathiques comme la variabilité de taille de fibres, des noyaux internalisés ainsi qu’un degré variable de nécrose et de fibrose endomysiale. Par ailleurs, il existe des anomalies de l’architecture myofibrillaire comme la présence de fibres lobulées, en tourbillon ou mitées, ainsi que des régions dépourvues d’activité oxydative qui ressemblent à des cores.
L’imagerie musculaire de ces patients montre souvent des anomalies qui ressemblent à celles typiquement associées aux COL6-RM [15,16], comme une atteinte très marquée du quadriceps débutant à la périphérie des fibres avec une préservation variable de la partie centrale du muscle. Au niveau du muscle droit fémoral, on peut observer l’image typique de cible ou target sign. Ces signes peuvent être aussi observés au sein de certains autres muscles tels que le moyen fessier ou le biceps fémoral. Par ailleurs, il existe une atteinte plus marquée des muscles de la ceinture pelvienne et du jambier antérieur, ce qui peut aider à les distinguer des COL6-RM [1]. Chez certains patients, l’imagerie montre une relative conservation du droit fémoral et du sartorius avec une distribution qui ressemble à celle de la dystrophie musculaire liée à POGLUT1 [17].
Jusqu’à présent, 15 variants pathogènes de JAG2 ont été rapportés : dix variants faux-sens, un variant non-sens, deux variants tronquants et deux délétions, tous compatibles avec une transmission autosomique récessive et une perte de fonction. Les mutations situées dans les exons 2 à 5 sont associées à une perte de la marche précoce (avant l’âge de 10 ans). L’analyse du transcriptome à partir de muscles de certains patients montre une diminution d’expression de facteurs impliqués dans la myogenèse comme PAX7 ou MYF5. La réduction d’expression (knock-down) de Jag2 dans une lignée de cellules murines conduit à une expression diminuée de Megf10, qui interagit avec la voie Notch et joue un rôle important dans le développement musculaire [1].
Le phénotype clinique se rapproche de celui des COL6-RM [18, 19] mais aussi d’autres entités comme la dystrophie des ceintures liée aux mutations récessives de POGLUT1 (LGMD R21) [17] ou à la myopathie congénitale liée aux mutations de MEGF10 (EMARDD : Early-Onset Myopathy, Areflexia, Respiratory Distress, and Dysphagia) [20], cette dernière associant un phénotype histologique similaire avec présence de cores. Néanmoins, l’atteinte respiratoire dans le cas des mutations JAG2 est souvent asymptomatique et moins sévère par rapport à la LGMD R21 ou à l’EMARDD. Concernant le diagnostic différentiel avec la COL6-RM, les rétractions dans cette dernière sont souvent plus marquées et associées souvent à une hyperlaxité distale et une atteinte cutanée (cicatrices chéloïdes, peau granuleuse). Il peut y avoir également une atteinte respiratoire variable mais pas d’atteinte cardiaque. Néanmoins, l’imagerie musculaire est très similaire dans les deux cas avec des signes très typiques comme le signe du sandwich ou du target. Dans le cas de JAG2, ces anomalies peuvent être observées aussi dans d’autres muscles en dehors du quadriceps comme le biceps fémoral ou les fessiers. Le signe de la cible au niveau du muscle droit fémoral est moins fréquemment retrouvé. Par ailleurs, d’autres myopathies doivent être considérées dans le diagnostic différentiel, notamment dans le cas de patients présentant un début précoce comme les myopathies liées aux mutations de SEPN1 (SEPN1-RM) [21] qui présentent une atteinte axiale et respiratoire précoce ou les myopathies liées à un déficit en mérosine (mutations du gène LAMA2) associées parfois à une épilepsie et, en imagerie cérébrale, à des anomalies de la substance blanche étendue [22]. D’autres maladies musculaires volontiers rétractiles (dystrophies musculaires, myopathies inflammatoires) peuvent être évoquées avec ou sans anomalies structurales du SNC [23] (Figure 3).
 |
Figure 3. Diagnostic différentiel. |
Concernant les mécanismes pathologiques impliqués, ces entités pourraient partager des mécanismes communs comme le dysfonctionnement de la voie Notch et/ou la déplétion de cellules satellites, impliqué aussi dans les myopathies liées aux mutations de PAX7 [24], POGLUT1 [17], MEGF10 [25] ou SEPN1 [26]. En ce sens, cette voie pourrait constituer un cible thérapeutique commune pour ces myopathies pour lesquelles il n’existe actuellement pas de traitement spécifique.
Liens d’intérêt
Les auteures déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Coppens S, Barnard AM, Puusepp S, et al. A form of muscular dystrophy associated with pathogenic variants in JAG2. Am J Hum Genet 2021; 108 : 840–56. [CrossRef] [PubMed] [Google Scholar]
- Luo B, Aster JC, Hasserjian RP, et al. Isolation and functional analysis of a cDNA for human Jagged2, a gene encoding a ligand for the Notch1 receptor. Mol Cell Biol 1997 ; 17 : 6057–6067. [CrossRef] [PubMed] [Google Scholar]
- Deng Y, Madan A, Banta AB, et al. Characterization, chromosomal localization, and the complete 30-kb DNA sequence of the human Jagged2 (JAG2) gene. Genomics 2000 ; 63 : 133–138. [CrossRef] [PubMed] [Google Scholar]
- Suckling RJ, Korona B, Whiteman P, et al. Structural and functional dissection of the interplay between lipid and Notch binding by human Notch ligands. EMBO J 2017 ; 36 : 2204–2215. [CrossRef] [PubMed] [Google Scholar]
- Luo D, Renault VM, Rando TA. The regulation of Notch signaling in muscle stem cell activation and postnatal myogenesis. Semin Cell Dev Biol 2005 ; 16 : 612–622. [CrossRef] [PubMed] [Google Scholar]
- Mourikis P, Tajbakhsh S. Distinct contextual roles for Notch signalling in skeletal muscle stem cells. BMC Dev Biol 2014; 14. doi:10.1186/1471-213X-14-2. [CrossRef] [PubMed] [Google Scholar]
- Mašek J, Andersson ER. The developmental biology of genetic notch disorders. Dev 2017 ; 144 : 1743–1763. [CrossRef] [PubMed] [Google Scholar]
- Li L, Krantz ID, Deng Y, Genin A, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for notch1. Nat Genet 1997 ; 16 : 243–251. [CrossRef] [PubMed] [Google Scholar]
- Grochowski CM, Loomes KM, Spinner NB. Jagged1 (JAG1): structure, expression, and disease associations. Gene 2016 ; 576 : 381–384. [CrossRef] [PubMed] [Google Scholar]
- Bauer RC, Laney AO, Smith R, et al. Jagged1 (JAG1) mutations in patients with tetralogy of Fallot or pulmonic stenosis. Hum Mutat 2010 ; 31 : 594–601. [CrossRef] [PubMed] [Google Scholar]
- Fischer-Zirnsak B, Segebrecht L, Schubach M, et al. Haploinsufficiency of the Notch ligand DLL1 Causes variable neurodevelopmental disorders. Am J Hum Genet 2019 ; 105 : 631–639. [CrossRef] [PubMed] [Google Scholar]
- Bulman MP, Kusumi K, Frayling TM, et al. Mutations in the human Delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat Genet 2000 ; 24 : 438–441. [CrossRef] [PubMed] [Google Scholar]
- Sullivan JM, Motley WW, Johnson JO, et al. Dominant mutations of the Notch ligand Jagged1 cause peripheral neuropathy. J Clin Invest 2020; 130 : 1506–12. [CrossRef] [PubMed] [Google Scholar]
- Chabriat H, Joutel A, Dichgans M, et al. Lancet Neurol 2009 ; 8 : 643–653. [CrossRef] [PubMed] [Google Scholar]
- Mercuri E, Lampe A, Allsop J, et al. Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscul Disord 2005 ; 15 : 303–310. [CrossRef] [PubMed] [Google Scholar]
- Mercuri E, Cini C, Pichiecchio A, et al. Muscle magnetic resonance imaging in patients with congenital muscular dystrophy and Ullrich phenotype. Neuromuscul Disord 2003 ; 13 : 554–558. [CrossRef] [PubMed] [Google Scholar]
- Servián-Morilla E, Takeuchi H, Lee T V, et al. A POGLUT 1 mutation causes a muscular dystrophy with reduced Notch signaling and satellite cell loss. EMBO Mol Med 2016 ; 8 : 1289–1309. 10.15252/emmm.201505815 [CrossRef] [PubMed] [Google Scholar]
- Bönnemann CG. The collagen VI-related myopathies: muscle meets its matrix. Nat Rev Neurol 2011 ; 7 : 379–390. [CrossRef] [PubMed] [Google Scholar]
- Briñas L, Richard P, Quijano-Roy S, et al. Early onset collagen VI myopathies: genetic and clinical correlations. Ann Neurol 2010 ; 68 : 511–520. [CrossRef] [PubMed] [Google Scholar]
- Logan CV, Lucke B, Pottinger C, et al. Mutations in MEGF10, a regulator of satellite cell myogenesis, cause early onset myopathy, areflexia, respiratory distress and dysphagia (EMARDD). Nat Genet 2011 ; 43 : 1189–1193. [CrossRef] [PubMed] [Google Scholar]
- Villar-Quiles RN, von der Hagen M, Métay C, et al. The clinical, histologic, and genotypic spectrum of SEPN1-related myopathy: a case series. Neurology 2020; 95 : e1512–27. [CrossRef] [PubMed] [Google Scholar]
- Sarkozy A, Foley AR, Zambon AA, et al. LAMA2-related dystrophies: clinical phenotypes, disease biomarkers, and clinical trial readiness. Front Mol Neurosci 2020; 13. doi:10.3389/fnmol.2020.00123. [PubMed] [Google Scholar]
- Muntoni F, Torelli S, Brockington M. Muscular dystrophies due to glycosylation defects. Neurotherapeutics 2008 ; 5 : 627–632. [CrossRef] [PubMed] [Google Scholar]
- Feichtinger RG, Mucha BE, Hengel H, et al. Biallelic variants in the transcription factor PAX7 are a new genetic cause of myopathy. Genet Med 2019 ; 21 : 2521–2531. [CrossRef] [PubMed] [Google Scholar]
- Saha M, Mitsuhashi S, Jones MD, et al. Consequences of MEGF10 deficiency on myoblast function and Notch1 interactions. Hum Mol Genet 2017 ; 26 : 2984–3000. [CrossRef] [PubMed] [Google Scholar]
- Castets P, Bertrand AT, Beuvin M, et al. Satellite cell loss and impaired muscle regeneration in selenoprotein N deficiency. Hum Mol Genet 2011 ; 20 : 694–704. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Biopsie musculaire (deltoïde). Les colorations hématoxyline-éosine (HE) et trichrome de Gomori (TG) montrent la présence de fibres musculaires de taille inégale avec légère augmentation du tissu conjonctif interstitiel. Certaines fibres montrent un remaniement de la structure et de rares noyaux internalisés. La technique oxydative NADH révèle la présence de fibres musculaires de taille inégale dont nombreuses présentent un aspect lobulé avec désorganisation de la structure. La coloration ATP 9,40 montre des fibres musculaires de taille inégale ainsi que la présence de fibres de petite taille. |
| Dans le texte | |
|
Figure 2. Imagerie musculaire (IRM corps entier). Séquences pondérées en T1. A. Coupe transversale au niveau thoracique montrant une atrophie des muscles paravertébraux ainsi qu’une involution graisseuse très importante au niveau des bras, particulièrement biceps et triceps brachiaux, avec une image d’atrophie « en doigts de gants ». B. Coupe transversale des cuisses montrant une atrophie et une involution graisseuse des deux loges mais qui débute dans la périphérie des muscles (aspect en « sandwich » sur le muscle vaste latéral). On note également le signe de la cible (target sign) sur le droit fémoral. C. Coupe transversale des jambes qui révèle une atrophie et un remplacement graisseux très importants au niveau des muscles gastrocnémiens latéral et médial et à moindre degré des muscles jambiers antérieurs, extenseurs des orteils et péroniers avec relative conservation du muscle tibial postérieur. |
| Dans le texte | |
|
Figure 3. Diagnostic différentiel. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.