")
")
| Issue |
Med Sci (Paris)
Volume 33, Number 12, Décembre 2017
|
|
|---|---|---|
| Page(s) | 1055 - 1062 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20173312012 | |
| Published online | 20 December 2017 | |
La modélisation mathématique, un outil essentiel pour l’étude du ciblage thérapeutique des tumeurs solides
Mathematical modeling: an essential tool for the study of therapeutic targeting in solid tumors
1
Laboratoire d’oncobiologie moléculaire, Centre de biologie humaine (CBH), CHU Amiens Sud, Amiens, France
2
Laboratoire amiénois de mathématique fondamentale et appliquée (LAMFA), CNRS UMR7352, UFR des sciences, Université de Picardie Jules Verne, Amiens, France
3
Laboratoire de biochimie, Centre de biologie humaine (CBH), CHU Amiens Sud, Amiens, France
4
Équipe CHIMERE (Chirurgie et extrémité céphalique, caractérisation morphologique et fonctionnelle), Université de Picardie Jules Verne, Amiens, France
a
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les progrès de la décennie passée ont rendu l’étude du traitement médical des cancers plus efficace, mais ils ont aussi révélé la complexité des mécanismes de la cancérogenèse. Pour la plupart des tumeurs humaines, on connaît plusieurs cibles thérapeutiques et des médicaments potentiels. Malheureusement, les acteurs identifiés comme des cibles thérapeutiques fonctionnent en effet souvent en réseaux, ce qui engendre des suppléances fonctionnelles et des propriétés de résilience/résistance au ciblage thérapeutique. Le recours à la modélisation mathématique permet de réaliser des études de système dans l’optique d’améliorer le ciblage thérapeutique des tumeurs solides. Nous présentons les principaux types de modèles mathématiques et des exemples récents illustrant leur intérêt en oncobiologie moléculaire.
Abstract
Recent progress in biology has made the study of the medical treatment of cancer more effective, but it has also revealed the large complexity of carcinogenesis and cell signaling. For many types of cancer, several therapeutic targets are known and in some cases drugs against these targets exist. Unfortunately, the target proteins often work in networks, resulting in functional adaptation and the development of resilience/resistance to medical treatment. The use of mathematical modeling makes it possible to carry out system-level analyses for improved study of therapeutic targeting in solid tumours. We present the main types of mathematical models used in cancer research and we provide examples illustrating the relevance of these approaches in molecular oncobiology.
© 2017 médecine/sciences – Inserm

Le cancer, une maladie complexe
Le cancer est une maladie complexe, caractérisée par l’occurrence d’anomalies allant du niveau moléculaire au corps entier. L’accumulation des mutations et les altérations touchant la régulation de l’expression de gènes contribuent à la transformation maligne [1]. L’issue de la maladie résulte des perturbations des systèmes qui régissent la transduction du signal dans la cellule cancéreuse, de la compétition entre cellules malignes et cellules saines, et de la capacité de la tumeur à coopter l’angiogenèse, à envahir les tissus et à former des métastases, et à échapper aux mécanismes effecteurs de l’immunité antitumorale [2]. Ces dernières décennies, les avancées de la génomique ont permis d’identifier des acteurs moléculaires essentiels de la cancérogenèse dont certains, comme les composants de la voie oncogénique RTK (receptor tyrosine kinase) – Ras – Raf – MEK (mitogen-activated ERK kinase) – ERK (extracellular signal-regulated kinase), constituent des cibles thérapeutiques potentielles [3].
La conception d’inhibiteurs spécifiques de ces protéines a permis de faire la preuve de principe de l’utilité de ces recherches avec, par exemple, le ciblage thérapeutique de B-Raf1 dans le mélanome malin [4]. Malheureusement, malgré un rationnel biologique établi, ces traitements anticancéreux présentent une efficacité qui se révèle le plus souvent temporaire. Après une période de temps limitée, des adaptations de la machinerie cellulaire peuvent apparaître et, dans certains cas, se traduire par l’inversion de l’effet du médicament, le transformant en un stimulant de la progression tumorale [5]. Des travaux récents suggèrent que ces remarquables propriétés d’adaptation peuvent découler de l’organisation intrinsèque des réseaux biologiques [6]. Il est donc nécessaire de prendre en compte l’organisation en réseaux et la régulation dynamique des cibles biologiques : c’est un des intérêts du recours à la modélisation mathématique [7–9] (→).
(→) Voir la Synthèse de Emmanuel Barillot et al., m/s n° 6-7, juin-juillet 2009, page 601
Nous présentons dans cette revue l’apport de la modélisation mathématique pour les études d’oncobiologie moléculaire et, plus précisément, pour l’étude de la transduction du signal dans la cellule cancéreuse.
Principaux modèles mathématiques en oncobiologie
Les étapes essentielles et les questions importantes pour la construction et l’utilisation des modèles mathématiques appliqués à l’étude du cancer, sont résumées dans la Figure 1. Schématiquement, les modèles mathématiques sont répartis selon la nature et le caractère prévisible des évènements qui sont modélisés, en modèles stochastiques ou en modèles déterministes [10]. Les modèles déterministes reposent sur l’utilisation d’équations différentielles ordinaires (EDO) et d’équations aux dérivées partielles (EDP). Les équations différentielles stochastiques (EDS) décrivent, elles, les évènements aléatoires. Les modèles mathématiques sont également différenciés en modèles continus et discrets. Les modèles continus sont adaptés à l’étude de la dynamique des phénomènes biologiques et donc, aux réponses des cellules cancéreuses dans le temps. Dans une optique formelle, les approches de modélisation sont souvent présentées selon deux catégories : (1) le « Bottom-up » : à partir d’éléments d’un système, une intégration des données est proposée afin de comprendre la fonctionnalité de l’ensemble. Ceci exige des connaissances préalables sur le système et nécessite que les interactions moléculaires et leurs paramètres cinétiques soient bien caractérisés. Les connaissances concernant les composants cellulaires sont ensuite fusionnées pour former un modèle plus large. Cette approche est particulièrement applicable à la biologie moléculaire et à la biochimie pour confirmer un comportement dans des cellules intactes, par exemple, l’organisation générale des grands réseaux de transduction oncogénique ; (2) le « Top-down », également connu sous le nom de reverse engineering. Cette approche convertit notamment les données issues du « big data » en connaissances sur un système. Elle est devenue plus courante avec la disponibilité accrue de données à grande échelle obtenues par les méthodes de métagénomique par exemple. Par la modélisation, le système est examiné afin de déterminer les principaux événements qui le régissent. Idéalement, la modélisation mathématique est conçue en parallèle d’expériences menées in vitro ou in vivo. Des allers-retours successifs entre ces deux approches (mathématique et biologique) permettent ainsi de construire et d’enrichir le modèle. Il sera validé lorsque les principales conclusions qui ont été émises in silico auront été confirmées expérimentalement. Il est important de mentionner que la réussite, comme l’échec, d’un modèle mathématique peuvent être informatifs. Il peut en effet être intéressant de révéler que les connaissances biologiques prises en compte pour la modélisation étaient insuffisantes. La modélisation n’est donc pas une fin en soi, mais une approche qui sera associée en parallèle, à des études de biologie [11] (→).
(→) Voir la Nouvelle de B. Martin et F. Mahé, page 1035 de ce numéro
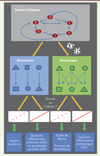 |
Figure 1. Les différents types de modèles mathématiques appliqués à l’oncobiologie. Pour construire un modèle mathématique d’un système biologique, il est important de définir les variables et l’échelle du modèle. Les principaux modèles utilisés pour étudier les systèmes biologiques se répartissent entre déterministe et stochastique. Dans les modèles déterministes, les résultats sont précisément déterminés par des relations connues entre les états et les événements, sans possibilité de variation aléatoire. Les équations différentielles ordinaires (EDO) et les équations aux dérivées partielles (EDP) ont pour inconnue une ou plusieurs fonctions, et sont adaptées aux analyses quantitatives du comportement d’un nombre limité d’acteurs biologiques. Au contraire, les modèles stochastiques sont une représentation mathématique d’un phénomène aléatoire, qui, même à partir de conditions initiales identiques, conduit à un ensemble de résultats différents à chaque simulation. Les processus de branchement constituent un type de processus de Markov, c’est-à-dire un type de processus stochastique possédant pour propriété essentielle que l’information utile pour prédire l’évolution du système modélisé est entièrement contenue dans son état présent. Les processus de branchement permettent d’étudier l’évolution dynamique d’une population de génération en génération. Ils sont typiquement utilisés pour modéliser l’accumulation des mutations et l’évolution des clones de cellules cancéreuses. À chaque cellule à un temps t est associée une probabilité intrinsèque de survenue d’un évènement (par exemple prolifération, mort cellulaire ou mutation). Par rapport aux EDO, les équations différentielles stochastiques (EDS) permettent une modélisation continue qui prend en compte le caractère stochastique de certains évènements. Alors que les modèles stochastiques sont plus représentatifs de la nature souvent aléatoire des réactions biologiques, ils sont algorithmiquement plus complexes et nécessitent un temps de calcul plus long par rapport aux modèles déterministes. Un compromis doit donc être trouvé entre simplicité/faisabilité et précision/meilleure représentation de la réalité. |
À l’heure actuelle, les équations différentielles ordinaires (EDO) représentent l’outil le plus largement utilisé pour les modélisations en biologie. Elles sont typiquement adaptées aux analyses quantitatives du comportement d’un nombre limité d’acteurs biologiques [12]. Une limitation importante à leur utilisation réside dans le fait qu’elles sont réservées à l’étude d’un petit nombre d’acteurs : les modèles reposant sur les EDO les plus larges incluent en effet une trentaine d’acteurs pour lesquels des données numériques sont disponibles [12]. D’autres formalismes mathématiques2 sont applicables lorsque les données ne sont pas adaptées aux EDO, par exemple lorsqu’il s’agit de données semi-quantitatives ou qualitatives.
Utilisation des équations différentielles ordinaires (EDO)
Les approches utilisées pour étudier les voies de signalisation intracellulaires reposent souvent sur l’application d’équations différentielles ordinaires (EDO) qui permettent d’en étudier la dynamique. Ces équations peuvent, par exemple, être construites à partir de la loi d’action de masse, ou de la cinétique de Michaelis-Menten, quand les interactions entre composants de ces voies sont de nature enzymatique [12]. La voie Raf-MEK-ERK est l’un des exemples de transduction oncogénique les plus étudiés sous le prisme mathématique, en raison de son implication fréquente dans le cancer [3, 9]. La première modélisation mathématique de cette voie oncogénique, centrée sur l’étude dynamique des composants Raf, MEK, et ERK, a été rapportée en 1996 [13]. Ce travail et plusieurs autres réalisés depuis, ont permis d’explorer des aspects importants de la régulation dynamique de cette cascade de transduction dans les cellules eucaryotes [12]. Parmi les contributions les plus notables, il a été possible d’expliquer (1) la cinétique d’activation hypersensible, en « tout ou rien », de ERK observée à l’échelle de la cellule unique [14], (2) d’explorer l’effet des protéines d’« échafaudage »3 [15], (3) d’expliquer les cinétiques distinctes d’activation de ERK selon les contextes de prolifération/différenciation cellulaire [16], ou encore (4) de mieux comprendre comment l’activation de ERK est convertie en régulation transcriptionnelle [17].
Nous avons étudié la voie Raf-MEK-ERK dans les cellules de carcinome hépatocellulaire (CHC) exposées au sorafénib4 [18] (Figure 2). Des EDO non-linéaires traduisant une cinétique enzymatique ont été utilisées pour modéliser les interactions, dans plusieurs lignées de CHC, entre les principaux acteurs de la voie Raf : B-Raf, C-Raf, MEK et ERK. Notre approche associant biologie et mathématiques a identifié deux points importants : (1) la grande hétérogénéité, selon les lignées de cellules, des modes de régulation de la signalisation en aval de Raf : l’analyse de sensibilité5 révèle, en effet, le rôle de régulateurs essentiels de la voie Raf, différents dans chacune des lignées ; (2) l’importance d’une étape insoupçonnée : l’étape de déphosphorylation des kinases ERK par les phosphatases. En l’absence de phosphatases spécifiques de MEK et de ERK, le sorafénib perd sa capacité de bloquer ERK dans des cellules pourtant sensibles à ce médicament [18]. Cette observation, quoique surprenante, le sorafénib ayant été conçu et développé comme inhibiteur de kinases, a pu être validée expérimentalement. Les approches de modélisation permettent donc de comprendre comment est régulée une voie oncogénique, comme la voie ERK, dans un contexte thérapeutique [18]. Ce type d’études permet secondairement, de guider la recherche de biomarqueurs originaux. Elles pourraient être utiles pour prédire l’efficacité du traitement et personnaliser la prescription du sorafénib. La modélisation mathématique révèle donc les détails de l’architecture en réseau des kinases Raf-MEK-ERK. Ces informations cruciales dans le contexte thérapeutique peuvent être obtenues par modélisation, même lorsque le réseau étudié est constitué d’un petit nombre d’acteurs (ici, les kinases Raf, MEK et ERK) [18, 19].
 |
Figure 2. Un exemple de modèle de la voie Raf-MEK-ERK reposant sur les équations différentielles ordinaires (EDO) et appliqué à l’étude du ciblage thérapeutique par le sorafénib dans le carcinome hépatocellulaire (CHC). A. La cascade Raf-MEK-ERK relaie les stimulus trophiques déclenchés par l’activation des RTK (récepteurs à activité tyrosine kinase) à la surface des cellules cancéreuses. B. Nous avons réalisé en western-blot une étude dynamique des niveaux de phosphorylation des kinases Raf, MEK et ERK dans les cellules de CHC exposées au sorafénib (appliqué à une concentration de 10 µm et maintenu à différents intervalles de temps). C. Pour chaque composant moléculaire du système, une réaction enzymatique d’activation par phosphorylation et une réaction inverse (déphosphorylation) sont décrites selon la cinétique de Michaelis-Menten avec des paramètres VM et KM (KM correspond à la valeur de la concentration de substrat pour laquelle la vitesse de réaction enzymatique est égale à la moitié de la vitesse maximale VM). Le système d’EDO correspondant est écrit et paramétré à partir des données expérimentales. D. Ce modèle permet d’étudier in silico la régulation de la voie Raf-MEK-ERK, en réalisant une analyse de sensibilité destinée à examiner l’importance relative de chaque paramètre dans le contrôle de la phosphorylation de ERK. Le modèle indique l’importance du paramètre VE,3, reflétant la réaction de disparition de ERK phosphorylée. E. Simulation de la cinétique de phosphorylation de ERK lorsque sont appliquées des variations sur le paramètre VE,3. On note que le modèle prédit l’absence complète d’effet inhibiteur du sorafénib sur la phosphorylation de ERK quand ce paramètre possède une valeur nulle (abolition de l’activité phosphatase de ERK). Cette prédiction a été vérifiée expérimentalement, permettant ainsi de valider le modèle et ses conclusions. μ* : moyenne absolue ; σ : déviation standard. Nous renvoyons le lecteur au travail original de Saidak et al. [18] pour plus de détails. |
Pour les réseaux complexes, qui peuvent inclure des milliers d’acteurs, il est nécessaire d’appliquer des stratégies de réduction de la complexité, comme l’analyse modulaire (modular response analysis, ou MRA). La première étape de ce type d’analyse repose sur l’identification des nœuds, c’est-à-dire des composants du système qui sont fortement connectés. Cette identification peut être fondée sur des connaissances antérieures du système, ou découler d’études statistiques (par exemple, par regroupement hiérarchique). La dynamique entre les nœuds peut ensuite être analysée à l’aide d’EDO. Pour chaque paramètre, un coefficient de réponse locale, qui reflète l’impact d’un nœud sur l’autre, est défini [20, 21]. Ce type d’analyses a été appliqué, par exemple, à la modélisation des interactions entre les voies oncogéniques Raf-MEK-ERK et AKT (ou protéine kinase B)-mTOR (mammalian target of rapamycin), dans des cellules de cancer colorectal. Elles ont permis de proposer un ciblage thérapeutique amélioré, en associant l’inhibition de MEK et de l’EGFR (epidermal growth factor receptor)6 [20]. Ce type d’approche permet donc d’étudier l’impact des multiples boucles de rétrocontrôle au sein de larges réseaux de transduction.
Modélisation stochastique des voies oncogéniques
Les exemples de modélisation des niveaux d’activation des kinases oncogéniques que nous avons présentés reposent sur des analyses de populations cellulaires. Ils font donc abstraction du comportement individuel des cellules. À l’échelle de la cellule, les voies oncogéniques, comme ERK, sont activées préférentiellement sous la forme de pics [22, 23], apparaissant de façon partiellement stochastique, avec une fréquence qui conditionne les réponses des cellules, comme la mitogenèse [22, 23]. L’état d’activation des composants d’amont (le « bruit », qui est une caractéristique des réseaux biologiques), mais aussi les différentes concentrations des acteurs de la cascade de signalisation, rendent impossible une modélisation mathématique entièrement déterministe. La modélisation de cette composante stochastique permet d’explorer les différences de régulation dynamique de Raf-MEK-ERK entre cellules, et d’en trouver l’origine. On peut ainsi montrer l’importance des différences qui existent entre cellules, de génomes identiques, et qui s’exercent en amont du module MEK-ERK [24, 25]. Cette notion d’hétérogénéité cellulaire intra-tumorale est importante. Elle conditionne en effet de nombreux aspects de la progression tumorale et la réponse des tumeurs aux traitements médicaux [26]. Les travaux de modélisation des voies oncogéniques à l’échelle des cellules uniques révèlent l’existence d’une hétérogénéité phénotypique d’origine non-génétique, inhérente à l’organisation des réseaux de transduction au sein des cellules cancéreuses [24, 25]. Les modèles qui incorporent une composante stochastique permettent donc de décrire, et de comprendre en partie, cette hétérogénéité phénotypique qui caractérise les tumeurs.
D’autres approches pour la modélisation de voies oncogéniques
Des approches utilisant la logique booléenne, permettent également d’étudier les réseaux de transduction dans les cellules cancéreuses [27–29]. Sur le principe, l’analyse quantitative est, dans ce type de modèle, remplacée par des variables binaires qui décrivent la fonctionnalité de chaque élément : l’activation de chaque acteur d’un réseau est soumise à différentes conditions logiques, et la stabilité des réseaux booléens dépend des connections qu’établissent les nœuds qui les constituent. Ce type de modèle présente l’avantage de permettre l’étude de nombreux acteurs, généralement choisis à partir de connaissances préexistantes dans le domaine. L’un des objectifs de ce type de modélisation est d’identifier les états attracteurs, présumés représenter les états stables du système modélisé. Ces états stables peuvent, par exemple, correspondre aux différents statuts du réseau oncogénique dans un contexte de ciblage thérapeutique. L’étude de la régulation de la protéine p537, un des déterminants de l’efficacité des chimiothérapies anti-tumorales, offre un bon exemple d’application de la modélisation logique en oncologie [27] (Figure 3). Les modèles booléens peuvent également permettre d’appréhender certains mécanismes à l’origine de la plasticité phénotypique observée dans les cellules cancéreuses. En définissant les états attracteurs dans un système booléen, il est possible d’aborder l’existence de plusieurs états métastables8 au sein d’un système biologique [29]. Une étude menée sur les cancers du poumon à petites cellules, offre un exemple récent d’utilisation d’un modèle booléen fondé sur l’état d’activation de 33 facteurs de transcription. Il a en effet été possible dans ce cas de mettre en évidence plusieurs états du réseau transcriptionnel et de les relier à l’émergence d’une hétérogénéité phénotypique dans le contexte thérapeutique [30].
 |
Figure 3. Modélisation logique de la régulation de p53. Le panneau A présente les composants du système et leurs interactions. Les niveaux de p53 (ou TP53, tumor protein 53) sont régulés par l’ubiquitine ligase Mdm2 (mouse double minute 2 homolog) qui catalyse la dégradation continue de p53. Inversement, p53 diminue les niveaux de Mdm2 nucléaire en inhibant sa translocation vers le noyau. Dans le même temps, p53 active la transcription du gène MDM2. Les règles logiques qui sont appliquées entre les différentes variables sont montrées dans le panneau A, avec les symboles Booléens et leur signification : !=NOT ; &=AND ; |=OR. Dans la forme la plus simple de logique, les variables prennent une valeur de 1 (= présent) ou 0 (= absent). Cependant, dans des modèles plus complexes, il est possible d’attribuer d’autres valeurs que 1. L’état actuel du système est décrit par un vecteur d’état, par exemple 0101 (c’est à dire : p53 = absent, Mdm2 cytoplasmique = présent, Mdm2 nucléaire = absent, lésions de l’ADN = présent). Les variables du système sont liées par des fonctions logiques. En appliquant ces fonctions, il est possible de déterminer l’évolution de l’état du système. Le panneau B présente l’évolution de l’état 0200, où trois évolutions sont possibles. En utilisant ce type de modèle logique, il est possible d’identifier l’état 0110 (p53 = absent, Mdm2 cytoplasmique = présent, Mdm2 nucléaire = présent, lésions de l’ADN = absent, souligné ici en jaune) comme un état stable. Il est aussi possible d’appliquer des perturbations afin d’explorer la robustesse du système, et son évolution dans différentes conditions initiales. Cet exemple est tiré des travaux de Abou-Jaoude et al. [27]. |
Des approches pratiques permettent de contourner une limitation essentielle des modèles booléens qui tendent à refléter grossièrement la réalité biologique. Une étude récente rapporte ainsi l’intérêt d’un formalisme à l’interface entre logique booléenne et EDO afin de modéliser la dynamique des réseaux [31]. Cette étude, qui révèle l’existence de différences individuelles entre des lignées cellulaires cancéreuses coliques, montre également que l’information contenue dans l’organisation en réseau prédit la sensibilité des cellules à un panel de médicaments de ciblage thérapeutique, indépendamment de l’analyse génomique [31]. Ce type de modélisation apporte donc une information utile à la compréhension des mécanismes qui rendent les cellules cancéreuses sensibles ou résistantes au ciblage thérapeutique.
Perspectives cliniques
Les études que nous relatons apportent la preuve de principe que la modélisation mathématique permet d’étudier le fonctionnement dynamique des voies de transduction oncogéniques, et que cette information est utile afin d’identifier des modalités de ciblage thérapeutique qui soient optimisées. Désormais, un objectif essentiel de la modélisation mathématique consiste en la conception d’outils qui permettront d’apprécier, pour chaque patient, cette organisation dynamique et de reconstruire in silico le réseau de transduction oncogénique qui lui est propre, ceci dans l’optique du développement d’une médecine personnalisée et prédictive [32]. Il est donc important, après l’étape préclinique, d’envisager désormais des études clinico-biologiques. Ceci suppose d’appliquer la modélisation établie dans les lignées de cellules, aux réseaux de transduction du signal présents dans les tumeurs des patients, à partir des données biologiques accessibles dans le contexte clinique. Ces données sont souvent limitées, notamment parce qu’elles nécessitent la réalisation de biopsies. Les mathématiques offrent des solutions pratiques à cette limitation. Une étude récente montre comment il est possible de tirer parti des stratégies mathématiques d’optimisation combinatoire9 pour exploiter les données de phospho-protéomique obtenues à partir de biopsies de glioblastomes [33] : associée aux connaissances préexistantes des réseaux d’interaction entre protéines, et avec un nombre limité de marqueurs analysés, la modélisation a permis de reconstruire in silico les voies de transduction activées dans ces tumeurs [33]. Un deuxième objectif essentiel consistera à valider le caractère prédictif des modèles mathématiques, en les comparant notamment aux analyses « classiques », centrées sur les éléments isolés constituant les réseaux modélisés [34].
À long terme, modéliser en clinique les voies de transduction oncogénique devrait contribuer à favoriser l’émergence de l’oncologie prédictive [32]. L’utilisation des modèles mathématiques permet l’intégration de données obtenues à différents niveaux. L’oncologie prédictive repose ainsi sur la conception de larges modèles multi-échelle, qui combinent la modélisation mathématique des voies oncogéniques, et celle décrivant les interactions entre clones cellulaires [35–38], avec des aspects spécifiques, comme la pharmacocinétique/pharmacodynamique des médicaments [39]. Au final, la modélisation mathématique constitue une piste prometteuse pour optimiser les recherches biologiques et faciliter la transposition en clinique des connaissances qui en sont issues.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Les auteurs remercient le comité de la Somme de la Ligue contre le Cancer, le CHU d’Amiens, et le Conseil régional des Hauts de France pour leur soutien à la recherche.
Une des trois protéines Raf. Les mutations touchant BRAF sont trouvées dans différents types de tumeurs.
Se dit d’un ensemble de règles et propriétés mathématiques qui sont utilisées dans un cadre précis.
Les protéines d’échafaudage sont considérées comme des réacteurs chimiques sur lesquels les molécules impliquées dans le transfert des signaux viennent se fixer simultanément avec l’orientation qui leur permet de se rencontrer et d’interagir.
Le sorafénib a une action bloquante sur les protéines C-Raf et B-Raf. Il est le médicament de référence pour le carcinome hépatocellulaire.
Permet d’identifier les paramètres et les variables d’entrée qui ont une forte influence sur les sorties d’un modèle.
La signalisation induite par l’EGFR implique PI3K (phosphatidyl inositol-1,4,5-trisphosphate 3-kinase) à l’origine de l’activation d’AKT et de mTOR.
La protéine p53 est l’un des plus importants suppresseurs de tumeur. Le gène TP53 qui la code est muté dans près de la moitié des cas de cancers humains.
En biologie, un état métastable est un état apparemment stable d’un système, mais qui est susceptible de changer rapidement sous l’effet d’une perturbation.
Un problème d’optimisation combinatoire consiste en la recherche d’une solution optimale dans un ensemble fini, mais éventuellement large, de solutions.
Références
- Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science 2015 ; 349 : 1483–1489. [Google Scholar]
- Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell 2010 ; 18 : 884–901. [CrossRef] [PubMed] [Google Scholar]
- Fey D, Matallanas D, Rauch J, et al. The complexities and versatility of the RAS-to-ERK signalling system in normal and cancer cells. Semin Cell Dev Biol 2016 ; 58 : 96–107. [CrossRef] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011 ; 364 : 2507–2516. [Google Scholar]
- Das Thakur M, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013; 494 : 251–255. [CrossRef] [PubMed] [Google Scholar]
- Jordan NV, Bardia A, Wittner BS, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature 2016 ; 537 : 102–106. [CrossRef] [PubMed] [Google Scholar]
- Barillot E, Calzone L, Zinovyev A. Biologie des systèmes appliquée aux cancers. Med Sci (Paris) 2009 ; 25 : 601–607. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Janes KA, Lauffenburger DA. Models of signalling networks: what cell biologists can gain from them and give to them. J Cell Sci 2013 ; 126 : 1913–1921. [Google Scholar]
- Kolch W, Halasz M, Granovskaya M, Kholodenko BN. The dynamic control of signal transduction networks in cancer cells. Nat Rev Cancer 2015 ; 15 : 515–527. [Google Scholar]
- Altrock PM, Liu LL, Michor F. The mathematics of cancer: integrating quantitative models. Nat Rev Cancer 2015 ; 15 : 730–745. [Google Scholar]
- Martin B, Mahé F. Un modèle mathématique pour l’étude des interactions bactériennes en biofilm Med Sci (Paris) 2017 ; 33 : 1035–1038. [Google Scholar]
- Orton RJ, Sturm OE, Vyshemirsky V, et al. Computational modelling of the receptor-tyrosine-kinase-activated MAPK pathway. Biochem J 2005 ; 392 : 249–261. [CrossRef] [PubMed] [Google Scholar]
- Huang CY, Ferrell JE Jr. Ultrasensitivity in the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA 1996 ; 93 : 10078–10083. [CrossRef] [Google Scholar]
- Ferrell JE Jr, Machleder EM. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science 1998 ; 280 : 895–898. [Google Scholar]
- Levchenko A, Bruck J, Sternberg PW. Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc Natl Acad Sci USA 2000 ; 97 : 5818–5823. [CrossRef] [Google Scholar]
- Sasagawa S, Ozaki Y, Fujita K, Kuroda S. Prediction and validation of the distinct dynamics of transient and sustained ERK activation. Nat Cell Biol 2005 ; 7 : 365–373. [CrossRef] [PubMed] [Google Scholar]
- Nakakuki T, Birtwistle MR, Saeki Y, et al. Ligand-specific c-Fos expression emerges from the spatiotemporal control of ErbB network dynamics. Cell 2010 ; 141 : 884–896. [CrossRef] [PubMed] [Google Scholar]
- Saidak Z, Giacobbi AS, Louandre C, et al. Mathematical modelling unveils the essential role played by cellular phosphatases in the inhibition of RAF-MEK-ERK signalling by sorafenib in Hepatocellular carcinoma cells. Cancer Lett 2017 ; 392 : 1–8. [Google Scholar]
- Jensen KJ, Moyer CB, Janes KA. Network architecture predisposes an enzyme to either pharmacologic or genetic targeting. Cell Syst 2016 ; 2 : 112–121. [CrossRef] [PubMed] [Google Scholar]
- Klinger B, Sieber A, Fritsche-Guenther R, et al. Network quantification of EGFR signaling unveils potential for targeted combination therapy. Mol Syst Biol 2013 ; 9 : 673. [Google Scholar]
- Halasz M, Kholodenko BN, Kolch W, Santra T. Integrating network reconstruction with mechanistic modeling to predict cancer therapies. Sci Signal 2016; 9 : ra114. [Google Scholar]
- Albeck JG, Mills GB, Brugge JS. Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol Cell 2013 ; 49 : 249–261. [CrossRef] [PubMed] [Google Scholar]
- Voliotis M, Perrett RM, McWilliams C, et al. Information transfer by leaky, heterogeneous, protein kinase signaling systems. Proc Natl Acad Sci USA 2014 ; 111 : E326–E333. [CrossRef] [Google Scholar]
- Ryu H, Chung M, Dobrzyn´ski M, et al. Frequency modulation of ERK activation dynamics rewires cell fate. Mol Syst Biol 2015 ; 11 : 838. [Google Scholar]
- Filippi S, Barnes CP, Kirk PD, et al. Robustness of MEK-ERK dynamics and origins of cell-to-cell variability in MAPK signaling. Cell Rep 2016 ; 15 : 2524–2535. [CrossRef] [PubMed] [Google Scholar]
- Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature 2013 ; 501 : 328–337. [CrossRef] [PubMed] [Google Scholar]
- Abou-Jaoudé W, Ouattara DA, Kaufman M. From structure to dynamics: frequency tuning in the p53-Mdm2 network I. Logical approach. J Theor Biol 2009 ; 258 : 561–577. [CrossRef] [Google Scholar]
- von der Heyde S, Bender C, Henjes F, et al. Boolean ErbB network reconstructions and perturbation simulations reveal individual drug response in different breast cancer cell lines. BMC Syst Biol 2014 ; 8 : 75. [PubMed] [Google Scholar]
- Abou-Jaoudé W, Traynard P, Monteiro PT, et al. Logical modeling and dynamical analysis of cellular networks. Front Genet 2016 ; 7 : 94. [PubMed] [Google Scholar]
- Udyavar AR, Wooten DJ, Hoeksema M, et al. Novel hybrid phenotype revealed in small cell lung cancer by a transcription factor network model that can explain tumor heterogeneity. Cancer Res 2017 ; 77 : 1063–1074. [Google Scholar]
- Eduati F, Doldàn-Martelli V, Klinger B, et al. Drug resistance mechanisms in colorectal cancer dissected with cell type-specific dynamic logic models. Cancer Res 2017 ; 77 : 3364–3375. [Google Scholar]
- Yankeelov TE, Quaranta V, Evans KJ, Rericha EC. Toward a science of tumor forecasting for clinical oncology. Cancer Res 2015 ; 75 : 918–923. [Google Scholar]
- Tuncbag N, Milani P, Pokorny JL, et al. Network modeling identifies patient-specific pathways in glioblastoma. Sci Rep 2016 ; 6 : 28668. [CrossRef] [PubMed] [Google Scholar]
- Fey D, Halasz M, Dreidax D, et al. Signaling pathway models as biomarkers: Patient-specific simulations of JNK activity predict the survival of neuroblastoma patients. Sci Signal 2015; 8 : ra130. [Google Scholar]
- Marusyk A, Tabassum DP, Altrock PM, et al. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 2014 ; 514 : 54–58. [CrossRef] [PubMed] [Google Scholar]
- Almendro V, Cheng YK, Randles A, et al. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep 2014 ; 6 : 514–527. [CrossRef] [PubMed] [Google Scholar]
- Chisholm RH, Lorenzi T, Lorz A, et al. Emergence of drug tolerance in cancer cell populations: an evolutionary outcome of selection, nongenetic instability, and stress-induced adaptation. Cancer Res 2015 ; 75 : 930–939. [Google Scholar]
- Bhang HE, Ruddy DA, Krishnamurthy Radhakrishna V, et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med 2015 ; 21 : 440–448. [CrossRef] [PubMed] [Google Scholar]
- Barbolosi D, Ciccolini J, Lacarelle B, et al. Computational oncology: mathematical modelling of drug regimens for precision medicine. Nat Rev Clin Oncol 2016 ; 13 : 242–254. [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Les différents types de modèles mathématiques appliqués à l’oncobiologie. Pour construire un modèle mathématique d’un système biologique, il est important de définir les variables et l’échelle du modèle. Les principaux modèles utilisés pour étudier les systèmes biologiques se répartissent entre déterministe et stochastique. Dans les modèles déterministes, les résultats sont précisément déterminés par des relations connues entre les états et les événements, sans possibilité de variation aléatoire. Les équations différentielles ordinaires (EDO) et les équations aux dérivées partielles (EDP) ont pour inconnue une ou plusieurs fonctions, et sont adaptées aux analyses quantitatives du comportement d’un nombre limité d’acteurs biologiques. Au contraire, les modèles stochastiques sont une représentation mathématique d’un phénomène aléatoire, qui, même à partir de conditions initiales identiques, conduit à un ensemble de résultats différents à chaque simulation. Les processus de branchement constituent un type de processus de Markov, c’est-à-dire un type de processus stochastique possédant pour propriété essentielle que l’information utile pour prédire l’évolution du système modélisé est entièrement contenue dans son état présent. Les processus de branchement permettent d’étudier l’évolution dynamique d’une population de génération en génération. Ils sont typiquement utilisés pour modéliser l’accumulation des mutations et l’évolution des clones de cellules cancéreuses. À chaque cellule à un temps t est associée une probabilité intrinsèque de survenue d’un évènement (par exemple prolifération, mort cellulaire ou mutation). Par rapport aux EDO, les équations différentielles stochastiques (EDS) permettent une modélisation continue qui prend en compte le caractère stochastique de certains évènements. Alors que les modèles stochastiques sont plus représentatifs de la nature souvent aléatoire des réactions biologiques, ils sont algorithmiquement plus complexes et nécessitent un temps de calcul plus long par rapport aux modèles déterministes. Un compromis doit donc être trouvé entre simplicité/faisabilité et précision/meilleure représentation de la réalité. |
| Dans le texte | |
|
Figure 2. Un exemple de modèle de la voie Raf-MEK-ERK reposant sur les équations différentielles ordinaires (EDO) et appliqué à l’étude du ciblage thérapeutique par le sorafénib dans le carcinome hépatocellulaire (CHC). A. La cascade Raf-MEK-ERK relaie les stimulus trophiques déclenchés par l’activation des RTK (récepteurs à activité tyrosine kinase) à la surface des cellules cancéreuses. B. Nous avons réalisé en western-blot une étude dynamique des niveaux de phosphorylation des kinases Raf, MEK et ERK dans les cellules de CHC exposées au sorafénib (appliqué à une concentration de 10 µm et maintenu à différents intervalles de temps). C. Pour chaque composant moléculaire du système, une réaction enzymatique d’activation par phosphorylation et une réaction inverse (déphosphorylation) sont décrites selon la cinétique de Michaelis-Menten avec des paramètres VM et KM (KM correspond à la valeur de la concentration de substrat pour laquelle la vitesse de réaction enzymatique est égale à la moitié de la vitesse maximale VM). Le système d’EDO correspondant est écrit et paramétré à partir des données expérimentales. D. Ce modèle permet d’étudier in silico la régulation de la voie Raf-MEK-ERK, en réalisant une analyse de sensibilité destinée à examiner l’importance relative de chaque paramètre dans le contrôle de la phosphorylation de ERK. Le modèle indique l’importance du paramètre VE,3, reflétant la réaction de disparition de ERK phosphorylée. E. Simulation de la cinétique de phosphorylation de ERK lorsque sont appliquées des variations sur le paramètre VE,3. On note que le modèle prédit l’absence complète d’effet inhibiteur du sorafénib sur la phosphorylation de ERK quand ce paramètre possède une valeur nulle (abolition de l’activité phosphatase de ERK). Cette prédiction a été vérifiée expérimentalement, permettant ainsi de valider le modèle et ses conclusions. μ* : moyenne absolue ; σ : déviation standard. Nous renvoyons le lecteur au travail original de Saidak et al. [18] pour plus de détails. |
| Dans le texte | |
|
Figure 3. Modélisation logique de la régulation de p53. Le panneau A présente les composants du système et leurs interactions. Les niveaux de p53 (ou TP53, tumor protein 53) sont régulés par l’ubiquitine ligase Mdm2 (mouse double minute 2 homolog) qui catalyse la dégradation continue de p53. Inversement, p53 diminue les niveaux de Mdm2 nucléaire en inhibant sa translocation vers le noyau. Dans le même temps, p53 active la transcription du gène MDM2. Les règles logiques qui sont appliquées entre les différentes variables sont montrées dans le panneau A, avec les symboles Booléens et leur signification : !=NOT ; &=AND ; |=OR. Dans la forme la plus simple de logique, les variables prennent une valeur de 1 (= présent) ou 0 (= absent). Cependant, dans des modèles plus complexes, il est possible d’attribuer d’autres valeurs que 1. L’état actuel du système est décrit par un vecteur d’état, par exemple 0101 (c’est à dire : p53 = absent, Mdm2 cytoplasmique = présent, Mdm2 nucléaire = absent, lésions de l’ADN = présent). Les variables du système sont liées par des fonctions logiques. En appliquant ces fonctions, il est possible de déterminer l’évolution de l’état du système. Le panneau B présente l’évolution de l’état 0200, où trois évolutions sont possibles. En utilisant ce type de modèle logique, il est possible d’identifier l’état 0110 (p53 = absent, Mdm2 cytoplasmique = présent, Mdm2 nucléaire = présent, lésions de l’ADN = absent, souligné ici en jaune) comme un état stable. Il est aussi possible d’appliquer des perturbations afin d’explorer la robustesse du système, et son évolution dans différentes conditions initiales. Cet exemple est tiré des travaux de Abou-Jaoude et al. [27]. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.