")
")
| Issue |
Med Sci (Paris)
Volume 33, Number 6-7, Juin-Juillet 2017
|
|
|---|---|---|
| Page(s) | 620 - 628 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20173306019 | |
| Published online | 19 July 2017 | |
Les microARN
Nouveaux acteurs dans la physiopathologie de la sclérose en plaques
MiRNAs: new actors in the physiopathology of multiple sclerosis
1
Département de biologie, École Normale Supérieure de Lyon, France
2
Laboratoire de biologie et de modélisation de la cellule, UMR5239/École Normale Supérieure de Lyon, UMS 344 Biosciences Lyon Gerland, université de Lyon, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
La sclérose en plaques (SEP) est une maladie auto-immune inflammatoire et démyélinisante dont l’étiologie reste mal connue. L’étude des microARN (miARN) apporte un éclairage nouveau sur les mécanismes physiopathologiques mis en jeu dans la SEP. Une signature miARN spécifique est observée tant au niveau cellulaire que dans les compartiments extracellulaires, chez les patients atteints de SEP, et dans le modèle de SEP développé chez l’animal, l’encéphalomyélite auto-immune expérimentale (EAE). Dans ce modèle, une association étroite a pu être mise en évidence entre la dérégulation des miARN et une modulation de la réponse immunitaire à l’origine d’un phénotype neuro-inflammatoire. La détection quantitative des miARN circulants a-t-elle une valeur diagnostique ? Les miARN seront-ils les nouveaux biomarqueurs de la maladie ? C’est à ces questions que cette revue tente de répondre.
Abstract
Multiple sclerosis (MS) is an auto-immune demyelinating disorder characterized by a chronic neuro-inflammatory process associated with an infiltration of the central nervous system (CNS) by autoreactive lymphocytes. The etiology of the disease remains unclear but the recent discovery of a dysregulated miRNA network in both cells and extracellular fluids of MS patients has brought new insights on the pathophysiological mechanisms involved in this disorder. miRNAs can induce a T cell polarization towards a pathological Th17 or Th1 phenotype and a deleterious activation of microglia, the CNS-resident macrophages. We provide here a review of the most recent data regarding miRNA dysregulation and pathophysiological roles in MS patients and in the animal model of MS, EAE (experimental autoimmune encephalomyelitis). Moreover, we discuss the putative clinical value of miRNAs as a novel biomarker and diagnostic tool for MS.
© 2017 médecine/sciences – Inserm
Vignette (Photo © Inserm - Pascal Heitzler).
La sclérose en plaques (ou SEP) est une maladie auto-immune chronique caractérisée essentiellement par une infiltration du système nerveux central (SNC) par des cellules du système immunitaire adaptatif. Parmi ces cellules, les lymphocytes T autoréactifs sont à l’origine d’une inflammation locale et d’une démyélinisation progressive des axones, pouvant entraîner un processus de dégénérescence neuronale secondaire. Différentes sous-populations de lymphocytes T sont anormalement retrouvées dans les lésions cérébrales, dont des cellules T CD4+, produisant de l’IFN(interféron)-γ et de l’IL(interleukine)-17, des lymphocytes T CD8+ et des lymphocytes Tγδ [1]. Ces lymphocytes, au profil pro-inflammatoire, contribuent à l’inflammation. D’autres lymphocytes T, les lymphocytes T régulateurs (Treg), permettent de la réduire. Chez les patients atteints de SEP, un dérèglement de cet équilibre entre réponse inflammatoire et immunorégulation favoriserait l’inflammation. De nombreuses études suggèrent en effet que la maturation et la fonction des lymphocytes Treg périphériques sont diminuées au cours de la SEP [2]. Il est probable que les lymphocytes B jouent également un rôle important dans la SEP, comme en témoigne la présence de bandes oligoclonales d’anticorps dans le liquide céphalo-rachidien (LCR) détectées par iso-électrofocalisation1 chez plus de 95 % des patients atteints de SEP [3]. Les lymphocytes B infiltrent le SNC dès l’apparition d’un syndrome cliniquement isolé (symptômes présentés par les patients ayant leur première poussée inflammatoire de type SEP – myélite, névrite optique, etc.-, que celle-ci donne lieu à un diagnostic SEP – nécessitant une deuxième poussée de même type – ou non) [3]. Les cellules résidentes du SNC, dont les cellules microgliales (les macrophages résidents du SNC), participent également aux processus inflammatoires en lien avec la progression des lésions du tissu nerveux. Du point de vue clinique, la SEP est associée à des perturbations motrices, sensitives et cognitives qui aboutissent à un handicap plus ou moins sévère selon les profils évolutifs. Classiquement, on distingue deux formes de la maladie : celle où alternent des périodes de rechutes et de rémissions (forme rémittente ou RRMS pour relapsing remitting multiple sclerosis) et celle dite progressive (PPMS pour primary progressive multiple sclerosis), qui suit parfois une forme rémittente (SPMS pour secondary progressive multiple sclerosis), et au cours de laquelle les patients acquièrent progressivement un handicap irréversible sans phase de rémission. Malgré des différences phénotypiques cliniques, la période qui s’écoule avant l’atteinte de certains niveaux d’incapacité et l’âge auquel ces niveaux sont atteints, sont similaires pour les patients présentant une PPMS ou une SPMS. En conséquence, des recommandations récentes ont proposé de regrouper la PPMS et la SPMS en une seule entité, dénommée « maladie progressive ».
Les microARN (miARN) sont des ARN de petite taille qui régulent l’expression de gènes cibles soit par la dégradation des ARN messagers codés par ces gènes, soit par l’inhibition directe de leur traduction ou par leur déadénylation2 [4] (Figure 1). Les précurseurs de miARN sont produits dans le noyau où ils subissent une étape de maturation avant d’être exportés dans le cytosol de la cellule. Ils sont alors clivés par l’enzyme Dicer et pris en charge par le complexe RISC (RNA-induced silencing complex). Les miARN associés au complexe RISC exercent ensuite leur action régulatrice dans le cytoplasme [5] (→).
(→) Voir la Nouvelle de C. Hartmann et al., m/s n° 10, octobre 2004, page 894
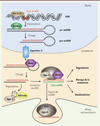 |
Figure 1. Biogenèse et mécanismes d’action des miARN. Les gènes codant les miARN sont transcrits par les ARN polymérases II et III. Les miARN formés, dits pri-miARN, sont pris en charge par un complexe comprenant la RNAse Drosha qui clive l’extrémité double-brin du pri-miARN, générant ainsi le pre-miARN. L’exportine-5 permet ensuite la translocation du pre-miARN vers le cytosol, où celui-ci est clivé par l’endonucléase Dicer. Un des brins, dit brin passager, est dégradé, probablement par l’action concertée d’hélicases et d’Ago2 (Argonaute 2), tandis que le brin complémentaire, fonctionnel, est chargé sur le complexe RISC (RNA-induced silencing complex) formé de Dicer, et d’autres protéines. L’association du complexe RISC et d’un miARN permet le ciblage de(s) l’ARN(s) messager(s) et conduit à l’inhibition de la traduction en protéine de l’ARN ciblé, (i) par dégradation de l’ARN messager, (ii) par blocage mécanique de la traduction (iii) ou par dénadénylation de l’ARN messager. Les miARN peuvent également être exportés vers d’autres cellules par l’intermédiaire de vésicules extracellulaires pour exercer leurs effets. |
Bien que de nombreux mécanismes moléculaires et cellulaires aient été décrits afin d’expliquer la physiopathologie de la SEP, l’étiologie de la maladie reste encore mal comprise. Au début des années 2000, l’importance des miARN a été décrite dans différents contextes pathologiques (le diabète [6] (→), les maladies cardiovasculaires [7, 58] (→) et le cancer [8]). Plusieurs équipes se sont alors intéressées à l’expression des miARN chez les patients atteints de SEP. D’autres ont étudié les polymorphismes des gènes codant ces miARN : des polymorphismes mononucléotidiques (ou SNP pour single nucleotide polymorphism) associés à la SEP ont ainsi été découverts dans certains gènes de miARN [9, 10]. Parallèlement, un vaste réseau de miARN, dont l’expression est modifiée chez les patients atteints de SEP, a été mis en évidence, principalement par des analyses génomiques à haut débit. Les miARN identifiés ont été isolés à partir de compartiments fonctionnels associés soit au système immunitaire soit au système nerveux. Ces miARN sont soit intracellulaires (isolés par extraction des miARN de cellules purifiées), soit extracellulaires (isolés à partir de fluides biologiques exempts de cellules). Cependant, en raison de l’importante quantité de données générées par ces analyses génomiques, l’étude fonctionnelle n’a été réalisée que pour quelques-uns de ces miARN.
(→) Voir la Synthèse de C. Hinault et al., m/s n° 8-9, août-septembre 2013, page 785
(→) Voir la Synthèse de F. Pinot et C. Bauters, m/s n° 8-9, août-septembre 2015, page 770
La plupart des données portant sur la fonctionnalité des miARN a été obtenue grâce à un modèle de SEP développé chez la souris, le modèle de l’encéphalomyélite auto-immune expérimentale ou EAE (experimental autoimmune encephalomyelitis). Ce modèle est fondé sur l’induction d’une réponse lymphocytaire T auto-immune dirigée contre la myéline, soit de façon active, par l’injection d’un broyat de moelle épinière ou de protéines purifiées de la myéline, soit de façon passive, par un transfert de lymphocytes T isolés d’animaux préalablement immunisés de façon active. Le plus souvent, l’EAE se caractérise par une paralysie, une perte de poids et un infiltrat de cellules mononucléées dans le SNC. Le degré de démyélinisation observé dépend du mode d’induction et du fond génétique des souches de souris utilisées.
Certains miARN, tels que le miR-155 ou le miR-326, semblent jouer un rôle majeur dans la physiopathologie de l’EAE, principalement en polarisant le système immunitaire vers une réponse inflammatoire qui se révèle délétère [11, 12]. Le fait que certains miARN extracellulaires soient également dérégulés dans la SEP suggère que leur transfert de cellule à cellule pourrait avoir un rôle physiopathologique. Les études pionnières, réalisées dans différents compartiments cellulaires et extracellulaires, ont permis d’établir des modèles montrant l’effet chronique de la dérégulation induite par les miARN au cours de la SEP. Enfin, il faut souligner que les études portant sur les miARN extracellulaires sont particulièrement importantes dans le cadre de la recherche de nouveaux biomarqueurs de la maladie.
Dérégulation des miARN chez les patients atteints de SEP
Au cours de ces dernières décennies, d’importantes avancées ont concerné les outils d’analyse permettant la détection et la quantification des ARN cellulaires codants ou non-codants. Il est désormais possible de quantifier simultanément des centaines de miARN grâce à des biopuces détectant des combinaisons choisies de miARN. Des matrices d’expression, selon les échantillons examinés, sont ainsi déduites à partir des miARN isolés et amplifiés.
L’utilisation de ces biopuces appliquée à différents tissus provenant de patients atteints de SEP a ainsi mis en évidence une altération de l’expression de nombreux miARN au cours de la maladie. La détection des miARN a été réalisée à partir de tissus (sang total [13, 14], tissu nerveux [15, 16], hippocampe [17]), de cellules (cellules mononucléées du sang [18–20], lymphocytes B [21–23] et T [11, 20, 23–26], monocytes [27], microglie [27], cellules endothéliales [28, 29]) ou de fluides extracellulaires (sérum/plasma [12, 18, 30–33], LCR [34]). Nous répertorions de manière systématique dans cette revue, l’ensemble des données issues de la littérature concernant les miARN dont l’expression apparaît modifiée chez les patients, par rapport à des individus sains. La Figure 2 , dans laquelle sont présentés leur localisation et leur nombre, illustre l’ampleur du réseau de miARN qui sont dérégulés dans la SEP, que ce soit dans le système périphérique sanguin (plus de 250 miARN) ou dans le SNC (plus de 100). Différents tissus, liquides physiologiques et types de cellules (immunitaires ou nerveuses) sont en effet concernés. Nous ne décrirons que certains miARN dont l’expression est altérée dans au moins trois compartiments (les miR-23a, 223, 22, 326, 21, 15a, 27a, 27b, 181c) ou qui ont une implication fonctionnelle dans la physiopathologie de la SEP (les miR-301a, 326, 155 et 21) (voir ci-après). Nous avons en effet considéré que le nombre de compartiments dans lesquels les miARN sont dérégulés était le reflet de l’étendue de la perturbation et de l’importance possible de ces miARN. Les miARN dont l’expression est altérée dans plusieurs compartiments seraient ainsi impliqués dans la régulation de voies cellulaires communes dans ces différents compartiments ou dans le contrôle qu’exerce un compartiment sur un autre.
 |
Figure 2. Les miARN sont dérégulés de façon généralisée chez les patients atteints de sclérose en plaques (SEP). Vingt-trois études répertoriées mettent en évidence la dérégulation de miARN chez des patients SEP par rapport à des individus sains. Ces miARN ont été classés selon leur compartiment de dérégulation, provenant soit du système périphérique sanguin (cadre supérieur), soit du sytème nerveux central (SNC, cadre inférieur). Les cadres gris rassemblent les miARN dérégulés dans le sang total (cadre supérieur) ou l’ensemble du SNC (cadre inférieur). Les cadres jaunes représentent la dérégulation de miARN dans les fluides. Le nom de certains miARN d’intérêt est mentionné pour chaque compartiment. |
Parmi les différents miARN identifiés, le miR-15a apparaît être un candidat particulièrement intéressant pour son implication potentielle dans la SEP. En effet, son expression chez les patients est modulée négativement dans quatre compartiments (le sang [14], les cellules mononucléées périphériques [20], les lymphocytes B [21] et T CD4+ [20]), et positivement dans deux autres (les lésions actives du tissu nerveux [15] et les lymphocytes Treg [26]) (Figure 2). D’autres critères sont également à prendre en considération pour sélectionner et valider les miARN ayant un rôle dans la SEP : (i) l’existence démontrée d’un ARN messager cible, (ii) l’ampleur de leur dérégulation et (iii) la validation de cette altération (évaluée par qRT-PCR [quantitative reverse transcription-polymerase chain reaction]). Ainsi, le miR-326 représente l’un des meilleurs candidats participant à la physiopathologie de la SEP avec une forte régulation positive dans trois compartiments différents [11, 12, 15] et l’existence d’un ARN messager cible, Ets-1 [V-ets avian erythroblastosis virus E26 oncogene homolog 1], démontrée in vivo [11].
La SEP est également étroitement associée à la dérégulation de miARN extracellulaires qui sont présents dans des liquides physiologiques comme le sérum, le plasma et le LCR (Figure 2). Étudier la façon dont les réseaux de miARN, cellulaires ou non-cellulaires, qui sont altérés dans la SEP, interagissent entre eux, constitue un important défi pour comprendre la physiopathologie de la maladie [35] (voir plus loin).
Rôle des miARN intracellulaires dans la SEP et l’EAE
L’expression de nombreux miARN, qui est altérée dans le SNC de patients atteints de SEP, l’est également dans le modèle murin d’EAE (miR-21 [15, 36], miR-326 [11, 15], miR-155 [12, 15], etc.) et les souris déficientes pour certains miARN (miR-21 -/- [36] et miR-155 -/- [37]) sont résistantes à l’induction d’une EAE. L’injection de mimétiques (c’est à dire de miARN synthétiques permettant de mimer l’action de miARN naturels) ou d’inhibiteurs de miARN, qu’ils soient intégrés à des lentivirus [11] ou associés à des agents de transfection [12], modifie la cinétique d’apparition des signes de la maladie, ce qui souligne encore l’importance des miARN dans le développement de l’EAE. Un rôle majeur de la différenciation des lymphocytes T naïfs en T pro-inflammatoires spécifiques de l’autoantigène myéline, de phénotype Th17 (produisant de l’IL-17) et Th1 (produisant de l’IFN-γ), a été démontré dans le développement de l’EAE [38]. Les miARN agissent en fait fréquemment sur la différenciation et/ou l’activation de ces cellules. Ainsi, lors de l’induction de l’EAE, la dérégulation de certains miARN (miR-301a [39], 326 [11], 155 [12], 21 [36]) qui modulent l’expression de facteurs contrôlant le statut des lymphocytes T (miR-301a, miR-21 et miR-326 inhibent respectivement PIAS3 [protein inhibitor of activated STAT (signal transducer and activator of transcription), 3] [39], SMAD [mothers against DPP homolog]-7 [36] et Ets-1 [11]), favorise l’émergence des phénotypes Th17 ou Th1 (Figure 3). Ainsi, altérer l’expression de ces miARN au cours de l’EAE provoque une augmentation du nombre de lymphocytes T pro-inflammatoires de types Th17 et Th1, ce qui accentue sa sévérité.
 |
Figure 3. Les miARN jouent un rôle pro-inflammatoire dans la physiopathologie de l’EAE (encéphalomyélite auto-immune expérimentale) et de la sclérose en plaques (SEP). Les miARN miR-301a, miR-21, miR-155 et miR-326 (marqués en gras) sont surexprimés dans le cadre de l’EAE et de la SEP. Les cellules T non différenciées (Th0) qui surexpriment ces miARN, se différencient préférentiellement en cellules Th1 (pour le miR-155) ou Th17 (pour les miR-155, miR-326, miR-21 et miR-301a). Les miARN régulent des voies majeures de différenciation cellulaire (TGF [transforming growth factor], STAT3 [signal transducer and activator of transcription 3], etc.). Les cellules Th1 et Th17 induites peuvent alors migrer dans le système nerveux central (SNC) et participer à l’inflammation. À ce niveau, la dérégulation de miR-155 et de miR-124 entraîne l’activation de la microglie, ce qui renforce l’inflammation et la démyélinisation des fibres nerveuses. Traits pleins : régulation effective ; Traits en tirets : régulation impossible (régulateur inhibé). |
Nous avons partagé jusqu’ici avec le lecteur une vision simplifiée de l’implication des lymphocytes T autoréactifs dans l’EAE. Le développement de l’EAE repose effectivement sur la différenciation de cellules Th17 et Th1 : les souris déficientes pour Rorγt (retinoic acid receptor-related orphan receptor gamma t, pour les cellules Th17) ou pour T-bet (T-box transcription factor, pour les Th1) sont résistantes à l’EAE [40,41]. En revanche les souris déficientes pour le gène Il-17 ou Ifn-g développent une EAE normale, voire plus sévère en ce qui concerne les souris déficientes pour l’Ifn-γ [42, 43]. Les lymphocytes Th17 et Th1 jouent donc bien un rôle primordial dans le développement de l’EAE. Cependant, le moyen par lequel ces cellules acquièrent leur pathogénicité n’est pas encore compris : la cellule T différenciée n’est pas délétère uniquement par la cytokine qu’elle produit (IL-17 pour Th17 et IFN-γ pour Th1). Il est donc cohérent que les miARN miR-301a, miR-21 et miR-326, participent au développement de l’EAE, puisqu’ils ciblent des facteurs gouvernant la différenciation des cellules en Th17 et pas uniquement la production des cytokines de type Th17 (IL-17, IL-22, etc.). Chez les patients atteints de SEP, différents éléments suggèrent pourtant un rôle pathologique de l’IL-17. En effet, ces patients présentent dans le sang et le LCR, des quantités plus importantes d’IL-17 que les sujets sains [44], ainsi qu’un infiltrat de lymphocytes Th17 dans le SNC [45]. Des thérapies ciblant l’IL-17, déjà approuvées dans le traitement d’autres maladies auto-immunes comme la spondylarthrite ankylosante et l’arthrite psoriasique, sont actuellement en essai clinique pour la SEP.
Les miARN peuvent également agir directement sur les cellules immunitaires résidentes du SNC. Ainsi, lors de l’induction de l’EAE, le niveau d’expression de miR-124 diminue au sein de la microglie, favorisant son activation [46] (Figure 3), ce qui impacte directement les processus de démyélinisation et d’inflammation au sein des lésions [47]. Le miR-155, qui est surexprimé lors de l’EAE, activerait également les cellules de la microglie et rendrait les cellules adjacentes susceptibles à la phagocytose par ces cellules [15].
Les miARN extracellulaires dans la SEP et l’EAE
L’implication des miARN dans la physiopathologie de la SEP et de l’EAE est fondée principalement sur l’étude des miARN intracellulaires. Pourtant, plusieurs observations récentes suggèrent des fonctions importantes des miARN extracellulaires. L’expression de nombreux miARN est en effet dérégulée chez les patients SEP, simultanément dans des compartiments intracellulaires et extracellulaires. Les miARN extracellulaires sont détectables dans la majorité des fluides biologiques (sérum, urine, LCR, salive, larmes, etc.) [48] et sont transportés par différents vecteurs moléculaires et cellulaires [49] (→). On retrouve en effet des miARN associés à des protéines circulantes ou inclus dans des vésicules comme les corps apoptotiques, les microvésicules et les exosomes. Les miARN extracellulaires participent-ils aux mécanismes physiopathologiques de la SEP impliquant les miARN intracellulaires ?
(→) Voir la Synthèse de S. Baulande et al., m/s n° 3, mars 2014, page 289
Les lymphocytes Th17 jouent un rôle important dans le développement de l’EAE et probablement de la SEP. Dans le modèle de l’EAE, la surexpression de miR-326 favorise une différenciation des lymphocytes T en lymphocytes Th17 [11]. Or, chez les patients SEP, ce miARN n’est pas surexprimé uniquement dans les lymphocytes T CD4+ [11] : on le retrouve également dans le sérum [12]. Les lymphocytes T produisent [50] et capturent [51] des vésicules extracellulaires qui permettent un transfert de molécules entre cellules. Un effet cellulaire de miR-326 pourrait donc, par transfert entre lymphocytes T, amplifier la différenciation cellulaire en Th17 (Figure 4). Cette hypothèse peut être envisagée pour d’autres situations. Le miR-155, par exemple, est surexprimé dans l’endothélium cérébral [29], dans le sérum [12] ainsi que dans les lésions cérébrales de patients SEP [15]. Or, son expression dans les cellules endothéliales module négativement les fonctions de la barrière hémato-encéphalique (avec une augmentation de sa perméabilité). Lopez-Ramirez et ses collègues ont en effet montré que la surexpression de miR-155 pouvait entraîner la dissociation de certains complexes de jonctions inter-endothéliales ou altérer les interactions cellule-matrice [29] (Figure 4). L’effet du transfert intercellulaire de miR-155 est l’une des hypothèses qui pourrait expliquer les mécanismes à l’origine d’une perméabilité prolongée de la barrière hémato-encéphalique chez les patients atteints de SEP (Figure 4). De même, O’Connell et ses collègues ont montré que le transfert de lymphocytes T de souris sauvages miR-155 +/+ chez des souris mutées miR-155 -/- suffisait pour restaurer le développement de l’EAE. Inversement, des souris miR-155 +/+ reconstituées avec la moelle osseuse provenant de souris déficientes miR-155 -/- deviennent résistantes à l’EAE. L’expression de miR-155 seulement dans les lymphocytes T semble donc conduire au développement de l’EAE [37]. Il reste à définir si la surexpression de miR-155 spécifiquement dans l’endothélium cérébral, en présence de lymphocytes T exprimant ce miARN normalement, peut jouer un rôle dans le développement de l’EAE et si l’origine de la surexpression de miR-155 dans l’endothélium cérébral est liée au transfert de vésicules issues des lymphocytes T.
 |
Figure 4. Modèle d’amplification des effets physiopathologiques des miARN extracellulaires. Le modèle présenté décrit le transfert intercellulaire de miARN par l’intermédiaire des vésicules extracellulaires. Ce modèle est cellulaire non-autonome puisqu’il impliquerait un collectif de cellules. Les exemples fournis sont ceux de miR-326, amplifiant la différenciation en Th17 et miR-155, augmentant la perméabilité de la barrière hémato-encéphalique. |
La mise en évidence de vésicules permettant le transfert de miARN fonctionnels dans la SEP pourrait aider à comprendre comment un déséquilibre immunitaire peut être à l’origine d’altérations du SNC. Dans un modèle murin, une étude récente suggère que, dans un contexte inflammatoire, de l’information génétique peut être transférée du compartiment immunitaire vers le SNC par l’intermédiaire de ces vésicules extracellulaires [52]. Cependant, le passage des vésicules issues de cellules immunitaires au travers de la barrière hémato-encéphalique et leur capture par des cellules du SNC reste à démontrer. Il est également important de définir l’origine de la dérégulation de miR-155 et de comprendre comment elle s’étend à d’autres compartiments (sérum [12], cellules endothéliales [29], monocytes [27], microglie [27], tissu nerveux lésé [15]).
L’atout « biomarqueur » des miARN dans la SEP
Malgré les progrès récents des approches thérapeutiques qui permettent de limiter la survenue des poussées chez les patients, la SEP reste à ce jour une maladie incurable. La découverte de nouveaux biomarqueurs moléculaires permettant d’optimiser la prise en charge de ces patients est donc un enjeu de premier ordre. Les miARN ont été décrits comme une nouvelle classe de biomarqueurs dans le cadre d’une médecine personnalisée [49] (→). Les miARN ont en effet l’avantage d’être accessibles rapidement et de façon peu invasive dans des biofluides comme le sang, l’urine ou les larmes. Ils sont aussi connus pour leur robustesse, stabilisés par leur vecteur et ils sont capables de résister à des conditions extrêmes de manipulation (variation de température, de pH, etc.) [53]. Par opposition aux méthodes actuelles de diagnostic comme l’imagerie par résonnance magnétique (IRM) ou l’étude du LCR par ponction lombaire, leur détection (par qPCR) est rapide et peu coûteuse.
(→) Voir la Synthèse de S. Baulande et al., m/s n° 3, mars 2014, page 289
Plusieurs études ont évalué l’utilisation des niveaux d’expression de miARN comme outil pour distinguer les patients atteints de SEP de sujets sains ou pour prédire les formes de SEP [18, 32, 34]. Les niveaux d’expression de certains miARN permettent en effet de diagnostiquer la SEP avec une précision de diagnostic supérieure à 70 % sur de petits échantillons. L’expression sérique de miR-223 et miR-15b permet par exemple une précision de diagnostic pour les patients PPMS de respectivement 80 et 75 % [32]. La valeur diagnostique des miARN comme biomarqueurs dépend essentiellement du nombre de patients et de sujets sains participant à l’analyse ainsi que l’utilisation combinée de plusieurs miARN. On peut néanmoins envisager que la détection des miARN à des temps précoces chez des sujets à risque puisse permettre, à terme, un dépistage de la SEP avant même l’apparition des premiers symptômes, mais aucune étude n’a, jusqu’à présent, fourni de valeur prédictive aux miARN. L’étude du profil d’expression des miARN dans le liquide cérébro-spinal est particulièrement pertinente puisqu’elle reflèterait le mieux les dommages précoces du cerveau. À ce jour, seuls Haghikia et ses collègues ont démontré que certains miARN sont dérégulés dans le LCR de patients atteints de SEP et que ces miARN pourraient être utilisés à des fins de diagnostic [34].
Dans l’optique du développement d’une médecine personnalisée et de traitements qui soient adaptés à chaque patient, une découverte intéressante a été que l’expression de miARN dérégulés dans la SEP peut être normalisée par la prise de certains médicaments. Des traitements (acétate de glatiramère, natalizumab, IFN-β) modifient en effet les niveaux d’expression de différents miARN (miR-146a [54], miR-142-3p [54], miR-126 [55] et miR-17 [56]). Chaque patient pourrait être ainsi suivi en fonction du profil de miARN préalablement prédéfinis pour différents compartiments et, selon ce profil ou son évolution, l’utilisation d’une combinaison de traitements, déjà existants, pourrait être proposée. La multithérapie ciblant les miARN spécifiquement dérégulés dans la SEP constitue donc une piste intéressante et innovante pour traiter les patients.
Conclusions et perspectives
Il est aujourd’hui démontré que les patients atteints de SEP présentent des anomalies d’expression de nombreux miARN, qu’ils soient intracellulaires ou extracellulaires. Ce processus est également observé dans le développement d’une EAE chez la souris. Dans ce modèle, la dérégulation des miARN favorise des mécanismes neuro-inflammatoires comme l’induction d’une réponse lymphocytaire de type Th17, la perméabilisation de la barrière hémato-encéphalique ou l’activation de la microglie. Cependant, l’origine de cette dérégulation dans la SEP demeure inconnue. Deux théories qui s’opposent l’une à l’autre, émergent. La première considère la dérégulation des miARN comme un des facteurs responsables de l’apparition de la SEP. À l’appui de cette théorie, la mise en évidence récente de l’association entre SEP et certains variants génétiques de miARN [9, 10]. La seconde théorie suppose que la dérégulation des miARN serait une conséquence d’un dérèglement préalable de la réponse neuro-immune. Dans cette hypothèse, la dérégulation des miARN participerait aux mécanismes pathologiques observés dans la SEP sans en expliquer l’étiologie.
À défaut de connaître la part de causalité des miARN dans la SEP, force est de constater leur importance dans le développement de l’EAE. Le modèle de l’EAE a beaucoup apporté à la compréhension des mécanismes physiopathologiques de la SEP. Il n’a cependant pas permis d’expliquer certains aspects de la maladie : histologique (la localisation des lésions) et cellulaire (l’importance clinique des réponses lymphocytaires B et T CD8 qui a été sous-estimée). En revanche, la dérégulation des miARN est un phénomène conservé entre SEP et EAE, suggérant son implication dans les réponses cellulaires à l’origine des atteintes cérébrales observées chez les patients atteints de SEP. Les vésicules extracellulaires, vecteurs des miRNA d’un compartiment vers un autre, pourraient également expliquer certains aspects, encore méconnus, des poussées inflammatoires qui caractérisent la maladie. Les transitions entre rémission et rechute sont ainsi corrélées à une augmentation d’expression de certains miARN [11,12]. Le modèle de l’effet cellulaire non-autonome des miARN décrit le processus par lequel les vésicules extracellulaires pourraient induire ou renforcer des changements d’expression de miARN intracellulaires. La concentration de vésicules extracellulaires, en particulier celles produites par les cellules endothéliales, serait en effet augmentée chez les patients en rechute [57], et susceptible de moduler la perméabilité de la barrière hémato-encéphalique [57]. Les miARN présents dans ces vésicules ont certainement un rôle à jouer mais il n’est pas encore clairement établi. Des innovations futures, en particulier dans le domaine des nanotechnologies, pourraient permettre d’identifier plus précisément l’impact de ces miARN extracellulaires dans le développement de la SEP.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
La présence d’immunoglobulines d’isotype G (IgG) dans le LCR, due à un excès de production d’IgG dans le système nerveux central, peut être révélée par la technique d’iso-électrofocalisation.
C’est-à-dire le raccourcissement de leur extrémité 5’ polyadénylée.
Références
- Fletcher JM, Lalor SJ, Sweeney CM, et al. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol 2010 ; 162 : 1–11. [Google Scholar]
- Zozulya AL, Wiendl H. The role of regulatory T cells in multiple sclerosis. Nat Clin Pract Neurol 2008 ; 4 : 384–398. [CrossRef] [PubMed] [Google Scholar]
- Disanto G, Morahan JM, Barnett MH, et al. The evidence for a role of B cells in multiple sclerosis. Neurology 2012 ; 78 : 823–832. [Google Scholar]
- Winter J, Jung S, Keller S, et al. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 2009 ; 11 : 228–234. [CrossRef] [PubMed] [Google Scholar]
- Hartmann C, Corre-Menguy F, Boualem A, et al. Les microARN: Une nouvelle classe de régulateurs de l’expression génique. Med Sci (Paris) 2004 ; 20 : 894–898. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Hinault C, Dumortier O, Van Obberghen E. MicroARN et diabète : petites structures - grands effets. Med Sci (Paris) 2013 ; 29 : 785–790. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- McManus DD, Freedman JE. MicroRNAs in platelet function and cardiovascular disease. Nat Rev Cardiol 2015 ; 12 : 711–717. [Google Scholar]
- Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer 2015 ; 15 : 321–333. [Google Scholar]
- Li Y, Du C, Wang W, et al. Genetic association of MiR-146a with multiple sclerosis susceptibility in the Chinese population. Cell Physiol Biochem 2015 ; 35 : 281–291. [CrossRef] [PubMed] [Google Scholar]
- Kiselev I, Bashinskaya V, Kulakova O, et al. Variants of MicroRNA genes: gender-specific associations with multiple sclerosis risk and severity. Int J Mol Sci 2015 ; 16 : 20067–20081. [Google Scholar]
- Du C, Liu C, Kang J, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol 2009 ; 10 : 1252–1259. [CrossRef] [PubMed] [Google Scholar]
- Zhang J, Cheng Y, Cui W, et al. MicroRNA-155 modulates Th1 and Th17 cell differentiation and is associated with multiple sclerosis and experimental autoimmune encephalomyelitis. J Neuroimmunol 2014 ; 266 : 56–63. [CrossRef] [PubMed] [Google Scholar]
- Keller A, Leidinger P, Steinmeyer F, et al. Comprehensive analysis of microRNA profiles in multiple sclerosis including next-generation sequencing. Mult Scler Houndmills Basingstoke Engl 2014 ; 20 : 295–303. [CrossRef] [Google Scholar]
- Cox MB, Cairns MJ, Gandhi KS, et al. MicroRNAs miR-17 and miR-20a inhibit T cell activation genes and are under-expressed in MS whole blood. PLoS One 2010 ; 5 : e12132. [CrossRef] [PubMed] [Google Scholar]
- Junker A, Krumbholz M, Eisele S, et al. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain J Neurol 2009 ; 132 : 3342–3352. [CrossRef] [Google Scholar]
- Noorbakhsh F, Ellestad KK, Maingat F, et al. Impaired neurosteroid synthesis in multiple sclerosis. Brain J Neurol 2011 ; 134 : 2703–2721. [CrossRef] [Google Scholar]
- Dutta R, Chomyk AM, Chang A, et al. Hippocampal demyelination and memory dysfunction are associated with increased levels of the neuronal microRNA miR-124 and reduced AMPA receptors. Ann Neurol 2013 ; 73 : 637–645. [CrossRef] [PubMed] [Google Scholar]
- Søndergaard HB, Hesse D, Krakauer M, et al. Differential microRNA expression in blood in multiple sclerosis. Mult Scler Houndmills Basingstoke Engl 2013 ; 19 : 1849–1857. [CrossRef] [Google Scholar]
- Otaegui D, Baranzini SE, Armañanzas R, et al. Differential micro RNA expression in PBMC from multiple sclerosis patients. PLoS One 2009 ; 4 : e6309. [CrossRef] [PubMed] [Google Scholar]
- Lorenzi JCC, Brum DG, Zanette DL, et al. miR-15a and 16–1 are downregulated in CD4+ T cells of multiple sclerosis relapsing patients. Int J Neurosci 2012 ; 122 : 466–471. [Google Scholar]
- Sievers C, Meira M, Hoffmann F, et al. Altered microRNA expression in B lymphocytes in multiple sclerosis: towards a better understanding of treatment effects. Clin Immunol Orlando Fla 2012 ; 144 : 70–79. [CrossRef] [Google Scholar]
- Miyazaki Y, Li R, Rezk A, et al. A Novel MicroRNA-132-surtuin-1 axis underlies aberrant b-cell cytokine regulation in patients with relapsing-remitting multiple sclerosis. PLoS One 2014 ; 9 : e105421. [CrossRef] [PubMed] [Google Scholar]
- Lindberg RLP, Hoffmann F, Mehling M, et al. Altered expression of miR-17-5p in CD4+ lymphocytes of relapsing-remitting multiple sclerosis patients. Eur J Immunol 2010 ; 40 : 888–898. [CrossRef] [PubMed] [Google Scholar]
- Jernås M, Malmeström C, Axelsson M, et al. MicroRNA regulate immune pathways in T-cells in multiple sclerosis (MS). BMC Immunol 2013 ; 14 : 32. [CrossRef] [PubMed] [Google Scholar]
- Guerau-de-Arellano M, Smith KM, Godlewski J, et al. Micro-RNA dysregulation in multiple sclerosis favours pro-inflammatory T-cell-mediated autoimmunity. Brain J Neurol 2011 ; 134 : 3578–3589. [CrossRef] [Google Scholar]
- De Santis G, Ferracin M, Biondani A, et al. Altered miRNA expression in T regulatory cells in course of multiple sclerosis. J Neuroimmunol 2010 ; 226 : 165–171. [CrossRef] [PubMed] [Google Scholar]
- Moore CS, Rao VTS, Durafourt BA, et al. miR-155 as a multiple sclerosis-relevant regulator of myeloid cell polarization. Ann Neurol 2013 ; 74 : 709–720. [CrossRef] [PubMed] [Google Scholar]
- Reijerkerk A, Lopez-Ramirez MA, van Het Hof B, et al. MicroRNAs regulate human brain endothelial cell-barrier function in inflammation: implications for multiple sclerosis. J Neurosci 2013 ; 33 : 6857–6863. [CrossRef] [PubMed] [Google Scholar]
- Lopez-Ramirez MA, Wu D, Pryce G, et al. MicroRNA-155 negatively affects blood-brain barrier function during neuroinflammation. FASEB J 2014 ; 28 : 2551–2565. [CrossRef] [PubMed] [Google Scholar]
- Gandhi R, Healy B, Gholipour T, et al. Circulating microRNAs as biomarkers for disease staging in multiple sclerosis. Ann Neurol 2013 ; 73 : 729–740. [CrossRef] [PubMed] [Google Scholar]
- Siegel SR, Mackenzie J, Chaplin G, et al. Circulating microRNAs involved in multiple sclerosis. Mol Biol Rep 2012 ; 39 : 6219–6225. [CrossRef] [PubMed] [Google Scholar]
- Fenoglio C, Ridolfi E, Cantoni C, et al. Decreased circulating miRNA levels in patients with primary progressive multiple sclerosis. Mult Scler Houndmills Basingstoke Engl 2013 ; 19 : 1938–1942. [CrossRef] [Google Scholar]
- Mancuso R, Hernis A, Agostini S, et al. MicroRNA-572 expression in multiple sclerosis patients with different patterns of clinical progression. J Transl Med 2015 ; 13 : 148. [CrossRef] [PubMed] [Google Scholar]
- Haghikia A, Haghikia A, Hellwig K, et al. Regulated microRNAs in the CSF of patients with multiple sclerosis: a case-control study. Neurology 2012 ; 79 : 2166–2170. [Google Scholar]
- Jagot F, Davoust N. Is it worth considering circulating microRNAs in multiple sclerosis ?. Front Immunol 2016 ; 7 : 129. [CrossRef] [PubMed] [Google Scholar]
- Murugaiyan G, da Cunha AP, Ajay AK, et al. MicroRNA-21 promotes Th17 differentiation and mediates experimental autoimmune encephalomyelitis. J Clin Invest 2015 ; 125 : 1069–1080. [CrossRef] [PubMed] [Google Scholar]
- O’Connell RM, Kahn D, Gibson WSJ, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 2010 ; 33 : 607–619. [CrossRef] [PubMed] [Google Scholar]
- El-behi M, Rostami A, Ciric B. Current views on the roles of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol 2010 ; 5 : 189–197. [Google Scholar]
- Mycko MP, Cichalewska M, Machlanska A, et al. MicroRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc Natl Acad Sci USA 2012 ; 109 : E1248–E1257. [CrossRef] [Google Scholar]
- Bettelli E, Sullivan B, Szabo SJ, et al. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med 2004 ; 200 : 79–87. [CrossRef] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006 ; 126 : 1121–1133. [CrossRef] [PubMed] [Google Scholar]
- Haak S, Croxford AL, Kreymborg K, et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest 2009 ; 119 : 61–69. [PubMed] [Google Scholar]
- Ferber IA, Brocke S, Taylor-Edwards C, et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol 1996 ; 156 : 5–7. [PubMed] [Google Scholar]
- Matusevicius D, Kivisäkk P, He B, et al. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler Houndmills Basingstoke Engl 1999 ; 5 : 101–104. [CrossRef] [EDP Sciences] [Google Scholar]
- Tzartos JS, Friese MA, Craner MJ, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol 2008 ; 172 : 146–155. [CrossRef] [PubMed] [Google Scholar]
- Ponomarev ED, Veremeyko T, Barteneva N, et al. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-α-PU.1 pathway. Nat Med 2011 ; 17 : 64–70. [CrossRef] [PubMed] [Google Scholar]
- Goldmann T, Prinz M. Role of microglia in CNS autoimmunity. J Immunol Res 2013 ; 2013 : e208093. [Google Scholar]
- Weber JA, Baxter DH, Zhang S, et al. The microRNA spectrum in 12 body fluids. Clin Chem 2010 ; 56 : 1733–1741. [CrossRef] [PubMed] [Google Scholar]
- Baulande S, Criqui A, Duthieuw M. Les microARN circulants, une nouvelle classe de biomarqueurs pour la médecine. Med Sci (Paris) 2014 ; 30 : 289–296. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Okoye IS, Coomes SM, Pelly VS, et al. MicroRNA-containing T-regulatory-cell-derived exosomes suppress pathogenic T helper 1 cells. Immunity 2014 ; 41 : 89–103. [CrossRef] [PubMed] [Google Scholar]
- Walker JD, Maier CL, Pober JS. Cytomegalovirus-infected human endothelial cells can stimulate allogeneic CD4+ memory T cells by releasing antigenic exosomes. J Immunol 2009 ; 182 : 1548–1559. [CrossRef] [PubMed] [Google Scholar]
- Ridder K, Keller S, Dams M, et al. Extracellular vesicle-mediated transfer of genetic information between the hematopoietic system and the brain in response to inflammation. PLoS Biol 2014 ; 12 : e1001874. [CrossRef] [PubMed] [Google Scholar]
- Hoy AM, Buck AH. Extracellular small RNAs: what, where, why ?. Biochem Soc Trans 2012 ; 40 : 886–890. [CrossRef] [PubMed] [Google Scholar]
- Waschbisch A, Atiya M, Linker RA, et al. Glatiramer acetate treatment normalizes deregulated microRNA expression in relapsing remitting multiple sclerosis. PLoS One 2011 ; 6 : e24604. [CrossRef] [PubMed] [Google Scholar]
- Meira M, Sievers C, Hoffmann F, et al. MiR-126: a novel route for natalizumab action ?. Mult Scler Houndmills Basingstoke Engl 2014 ; 20 : 1363–1370. [CrossRef] [Google Scholar]
- Meira M, Sievers C, Hoffmann F, et al. Unraveling natalizumab effects on deregulated miR-17 expression in CD4+ T cells of patients with relapsing-remitting multiple sclerosis. J Immunol Res 2014 ; 2014 : 897249. [CrossRef] [PubMed] [Google Scholar]
- Sáenz-Cuesta M, Osorio-Querejeta I, Otaegui D. Extracellular vesicles in multiple sclerosis: What are they telling us ?. Front Cell Neurosci 2014 ; 8 : 100. [PubMed] [Google Scholar]
- Pinet F. BautersC. Potentiel des ARN non codants comme biomarqueurs dans l’insuffisance cardiaque. Med Sci (Paris) 2015 ; 31 : 770–776. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Biogenèse et mécanismes d’action des miARN. Les gènes codant les miARN sont transcrits par les ARN polymérases II et III. Les miARN formés, dits pri-miARN, sont pris en charge par un complexe comprenant la RNAse Drosha qui clive l’extrémité double-brin du pri-miARN, générant ainsi le pre-miARN. L’exportine-5 permet ensuite la translocation du pre-miARN vers le cytosol, où celui-ci est clivé par l’endonucléase Dicer. Un des brins, dit brin passager, est dégradé, probablement par l’action concertée d’hélicases et d’Ago2 (Argonaute 2), tandis que le brin complémentaire, fonctionnel, est chargé sur le complexe RISC (RNA-induced silencing complex) formé de Dicer, et d’autres protéines. L’association du complexe RISC et d’un miARN permet le ciblage de(s) l’ARN(s) messager(s) et conduit à l’inhibition de la traduction en protéine de l’ARN ciblé, (i) par dégradation de l’ARN messager, (ii) par blocage mécanique de la traduction (iii) ou par dénadénylation de l’ARN messager. Les miARN peuvent également être exportés vers d’autres cellules par l’intermédiaire de vésicules extracellulaires pour exercer leurs effets. |
| Dans le texte | |
|
Figure 2. Les miARN sont dérégulés de façon généralisée chez les patients atteints de sclérose en plaques (SEP). Vingt-trois études répertoriées mettent en évidence la dérégulation de miARN chez des patients SEP par rapport à des individus sains. Ces miARN ont été classés selon leur compartiment de dérégulation, provenant soit du système périphérique sanguin (cadre supérieur), soit du sytème nerveux central (SNC, cadre inférieur). Les cadres gris rassemblent les miARN dérégulés dans le sang total (cadre supérieur) ou l’ensemble du SNC (cadre inférieur). Les cadres jaunes représentent la dérégulation de miARN dans les fluides. Le nom de certains miARN d’intérêt est mentionné pour chaque compartiment. |
| Dans le texte | |
|
Figure 3. Les miARN jouent un rôle pro-inflammatoire dans la physiopathologie de l’EAE (encéphalomyélite auto-immune expérimentale) et de la sclérose en plaques (SEP). Les miARN miR-301a, miR-21, miR-155 et miR-326 (marqués en gras) sont surexprimés dans le cadre de l’EAE et de la SEP. Les cellules T non différenciées (Th0) qui surexpriment ces miARN, se différencient préférentiellement en cellules Th1 (pour le miR-155) ou Th17 (pour les miR-155, miR-326, miR-21 et miR-301a). Les miARN régulent des voies majeures de différenciation cellulaire (TGF [transforming growth factor], STAT3 [signal transducer and activator of transcription 3], etc.). Les cellules Th1 et Th17 induites peuvent alors migrer dans le système nerveux central (SNC) et participer à l’inflammation. À ce niveau, la dérégulation de miR-155 et de miR-124 entraîne l’activation de la microglie, ce qui renforce l’inflammation et la démyélinisation des fibres nerveuses. Traits pleins : régulation effective ; Traits en tirets : régulation impossible (régulateur inhibé). |
| Dans le texte | |
|
Figure 4. Modèle d’amplification des effets physiopathologiques des miARN extracellulaires. Le modèle présenté décrit le transfert intercellulaire de miARN par l’intermédiaire des vésicules extracellulaires. Ce modèle est cellulaire non-autonome puisqu’il impliquerait un collectif de cellules. Les exemples fournis sont ceux de miR-326, amplifiant la différenciation en Th17 et miR-155, augmentant la perméabilité de la barrière hémato-encéphalique. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.