")
")
| Issue |
Med Sci (Paris)
Volume 33, Number 5, Mai 2017
|
|
|---|---|---|
| Page(s) | 480 - 483 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20173305007 | |
| Published online | 14 June 2017 | |
PiT1 : du transport de phosphate à la signalisation insulinique
PiT1: from phosphate transport to insulin signaling
1
Inovarion, F-75013, Paris, France
2
Inserm U1151-CNRS UMR8253, Institut Necker-Enfants Malades (INEM), université Paris Descartes, F-75993 Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
L’obésité est aujourd’hui une maladie répandue dans le monde entier. Selon l’Organisation Mondiale de la Santé, en 2014, 39 % des adultes de plus de 18 ans étaient en surpoids et, parmi eux, 13 % étaient obèses. Ces chiffres ont été multipliés par deux depuis 1980.
L’obésité est un facteur de risque aggravant pour des maladies cardiovasculaires, le diabète de type II et le syndrome métabolique [1] (→). Ce dernier se caractérise notamment par une augmentation du tissu adipeux viscéral, par une résistance à l’insuline et une hypertension.
(→) Voir la Synthèse de D. Junquero et Y. Rival, m/s n° 12, décembre 2005, page 1045
Le foie, organe majeur du métabolisme
Le foie est l’organe majeur qui régule à la fois l’homéostasie glucidique et lipidique. Chez un individu sain, l’apport de glucose par l’alimentation stimule la production d’insuline au niveau du pancréas. L’insuline agit sur différents tissus cibles afin de diminuer la glycémie sanguine et la ramener à un taux basal. Elle stimule l’entrée de glucose au niveau des muscles, du foie et du tissu adipeux (TA) où il va être stocké sous forme de lipides via la lipogenèse (dans le foie et le TA) ou sous forme de glycogène (dans le foie et le muscle). L’insuline inhibe également la production de glucose par le foie (appelée néoglucogenèse). Chez un individu insulino-résistant, les organes cibles ne répondent pas, ou mal, à l’insuline avec pour conséquence, un relargage continu d’acide gras par les adipocytes (hyperlipidémie), une production continue de glucose par le foie (hyperglycémie) ainsi qu’une accumulation d’acides gras sous forme de triglycérides (stéatose hépatique) [2].
Il était une « foie » PiT1…
La protéine membranaire PiT1 (inorganic phosphate transporter 1) est constituée de 12 domaines transmembranaires et d’une large boucle intracellulaire. Initialement identifiée comme un récepteur de rétrovirus, PiT1 joue un rôle de co-transporteur sodium-phosphate. Depuis quelques années, de nouvelles fonctions ont été mises en évidence pour cette protéine. Elle a été notamment impliquée dans la régulation de la prolifération, de l’adhérence et de la densité cellulaire, mais aussi dans l’apoptose induite par le TNF-α (tumor necrosis factor alpha) ou encore la différenciation érythrocytaire [3]. La plupart de ces nouvelles fonctions ont été décrites comme étant indépendantes de sa fonction de transporteur de phosphate. L’invalidation totale du gène codant la protéine PiT1 chez la souris a pour conséquence une létalité embryonnaire à 12,5 jours de gestation. Les embryons meurent d’une anémie sévère et présentent un foie apoptotique, celui-ci étant l’organe hématopoïétique à ce stade du développement [4].
Dans le but de déterminer si la protéine PiT1 pouvait jouer un rôle dans le fonctionnement du foie, nous avons croisé des souris portant le gène PiT1 flanqué de 2 sites loxP1 avec des souris exprimant la recombinase Cre1 sous contrôle du promoteur de l’albumine [5]. Ce promoteur, exprimé uniquement dans les hépatocytes, n’est que très faiblement actif en fin de gestation, son activité augmentant ensuite graduellement après la naissance [6] . Les souris qui naissent de ce croisement (que nous nommerons par la suite souris TG pour transgéniques) sont donc invalidées pour le gène PiT1 spécifiquement au niveau des hépatocytes et atteignent l’âge adulte.
L’invalidation hépatocytaire de PiT1 conduit à un phénotype métabolique
Nos premières observations [5] montrant que les souris TG prenaient moins de poids que les souris contrôles (CT), nous ont conduit à nous intéresser à leur métabolisme. Nous avons mis en évidence que cette prise de poids diminuée s’accompagnait d’une masse de TA plus faible. Ces différences de prise de poids ne proviennent pas d’un déséquilibre entre l’apport alimentaire et les dépenses énergétiques. La mesure du quotient respiratoire indique toutefois que les souris TG ont une préférence pour l’utilisation des carbohydrates comme substrat énergétique (vs l’utilisation des lipides). Ceci pourrait, en partie, expliquer l’hypoglycémie à jeun observée chez ces animaux. Un défaut dans la production de glucose hépatique pourrait également être à l’origine de cette hypoglycémie à jeun. En effet, alors que le glucose est stocké sous forme de glycogène en période nourrie, il est produit par le foie via la néoglucogenèse au cours du jeûne. Afin de tester cette hypothèse, nous avons évalué les niveaux d’expression d’enzymes clés de la néoglucogenèse et, de façon surprenante, nous avons observé une augmentation de l’expression du co-activateur Pgc1α (PPARγ [peroxisome proliferator-activated receptor γ] coactivator 1α) et de l’enzyme PEPCK (phosphoenolpyruvate carboxykinase) dans le foie des souris TG. En revanche, la production de glucose à partir du pyruvate était équivalente pour les deux génotypes, à la fois in vitro, dans des hépatocytes primaires en culture, et in vivo. Ces résultats indiquent que la production de glucose hépatique n’est pas altérée par l’absence de la protéine PiT1 et suggèrent que l’augmentation d’expression des enzymes de la néoglucogenèse que l’on observe survient en réponse à l’hypoglycémie à jeun chez les souris TG.
PiT1 intervient dans le rétrocontrôle négatif de la signalisation insulinique
Sensibilisation à l’insuline in vivo
L’insuline est l’hormone qui régule la glycémie. Sa liaison à ses récepteurs induit leur autophosphorylation ainsi que la phosphorylation de substrats en aval, notamment l’IRS1 (insulin receptor substrat 1) [7] (→). Les tyrosines phosphorylées de l’IRS1 sont des sites de liaison pour la kinase PI3K (phosphatidylinositol 3-kinase), dont l’activation conduira à celle de la kinase AKT (aussi appelée protéine kinase B) (Figure 1) [8]. Nous avons observé que les souris TG, à l’état nourri, présentaient une glycémie équivalente à celle des souris CT, mais que leur insulinémie associée était plus basse. Des tests de tolérance au glucose et à l’insuline ont mis en évidence, chez les souris TG, une meilleure tolérance au glucose, ainsi qu’une meilleure sensibilité à l’insuline. Une augmentation de la signalisation insulinique, se manifestant par une phosphorylation plus importante du récepteur à l’insuline, d’AKT et de GSK3 (glycogen synthase kinase 3), a également été observée in vivo.
(→) Voir la Synthèse de J. Capeau, m/s n° 8-9, août-septembre 2003, page 834
 |
Figure 1. Représentation schématique de la voie de signalisation insulinique. La liaison de l’insuline à son récepteur entraîne l’autophosphorylation de celui-ci et son activation, qui induit le recrutement de protéines d’ancrage telles que l’IRS1 (insulin receptor substrat 1). L’IRS1 va être phosphorylée à son tour, ce qui va permettre le recrutement de la PI3K (phosphatidylinositol 3-kinase). Celle-ci phosphoryle les phosphatidylinositol diphosphates (PIP2) de la membrane cellulaire en phosphatidylinositol triphosphates (PIP3), activant ainsi AKT2. Cette dernière va exercer diverses actions : stimuler la synthèse de lipides et du glycogène, inhiber la production de glucose par le foie et favoriser l’entrée de glucose dans les cellules. PTEN (phosphatase and tensin homolog) possède un rôle antagoniste à la PI3K et inhibe l’activation d’AKT. Les phosphorylations sur des tyrosines sont représentées par des cercles rouges. Foxo : forkhead box O ; GSK3 : glycogen synthase kinase 3 ; SREBP : sterol regulatory element binding protein ; AKT2 : PKB (protéine kinase B) bêta. |
Prolongation du signal insulinique in vitro
Afin de mieux comprendre les mécanismes impliqués, nous avons étudié la signalisation insulinique in vitro dans des fibroblastes embryonnaires de souris (MEF) sauvages (CT) et invalidés pour le gène PiT1 (KO, knock-out). Nous avons observé, dans les MEF KO, un taux plus important de récepteurs de l’insuline ainsi qu’une phosphorylation d’AKT prolongée dans le temps. Parallèlement, nous avons mis en évidence une cinétique plus lente de dégradation de l’IRS1, cette dégradation constituant l’un des systèmes de régulation qui permet de stopper la signalisation insulinique [9]. À la suite de la liaison de l’insuline à son récepteur, l’IRS1 des MEF sauvages est phosphorylé sur des sérines (S) / thréonines (T), entraînant son ubiquitination et sa dégradation par le protéasome [10]. Dans les MEF KO, on observe une absence de phosphorylation des S/T de l’IRS1, ainsi qu’une diminution de son ubiquitination après stimulation par l’insuline. L’IRS1 est ainsi stabilisé, entraînant un retard dans le rétrocontrôle négatif de la signalisation insulinique dans les cellules MEF KO.
La protéine USP7, le lien entre PiT1 et IRS1
Parmi les partenaires possibles de la protéine PiT1, identifiés au cours d’un criblage double hybride chez la levure2, nous nous sommes intéressés plus particulièrement à la protéine USP7 (ubiquitin-specific-processing protease 7), une dé-ubiquitinase connue pour se lier à l’IRS1 et empêcher sa dégradation [11]. Des expériences de co-immunoprécipitation ont montré que les protéines USP7 et IRS1 interagissent dans les cellules MEF sauvages et KO en condition de privation. L’ajout d’insuline dans le milieu entraîne la dissociation du complexe USP7-IRS1 dans les cellules MEF CT, alors que les deux protéines restent liées dans les MEF KO. Par ailleurs, PTEN (phosphatase and tensin homolog) une tyrosine phosphatase de l’IRS1 [12] connue pour interagir, elle aussi, avec USP7 [13], est plus faiblement associée à l’IRS1 dans les MEF KO en réponse à l’insuline. L’ensemble de ces données suggèrent que la protéine PiT1 module les interactions USP7/IRS1 et PTEN/IRS1, jouant ainsi un rôle sur l’ubiquitination et la dégradation de l’IRS1. L’expression de novo de PiT1 (sauvage ou mutée pour sa fonction de transporteur de phosphate) dans des cellules MEF KO restaure le phénotype sauvage, confirmant son rôle dans la signalisation insulinique et l’ubiquitination de l’IRS1. La suppression de USP7 à l’aide de siARN (small interfering RNA) restaure également le phénotype, suggérant ainsi que la protéine PiT1 module la signalisation insulinique via son interaction avec USP7 (Figure 2).
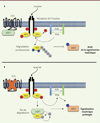 |
Figure 2. Représentation schématique de l’effet de l’invalidation de PiT1 (inorganic phosphate transporter 1) sur la signalisation insulinique. (A) En présence de PiT1, un rétrocontrôle négatif, déclenché par un signal insulinique prolongé, va permettre l’arrêt du signal. PTEN (phosphatase and tensin homolog) déphosphoryle le phosphatidylinositol triphosphate PIP3 en phosphatidylinositol diphosphate PIP2 et, ainsi, inactive la kinase AKT2. PTEN déphosphoryle également les tyrosines de l’IRS1 (insulin receptor substrat 1), ce qui libère la PI3K (phosphatidylinositol 3-kinase). De plus, le complexe USP7 (ubiquitin-specific-processing protease 7)/IRS1 se dissocie et entraîne l’ubiquitination (Ub, ubiquitine) de l’IRS1 et sa dégradation par le protéasome. (B) En l’absence de PiT1, l’expression de PTEN et son association avec l’IRS1 diminue, n’entraînant pas l’inactivation d’AKT2. De plus, USP7 reste associée à l’IRS1, la protégeant ainsi de l’ubiquitination et empêchant donc sa dégradation. Les phosphorylations sur des sérines/thréonines sont représentées par des cercles bleus. Les phosphorylations sur des tyrosines sont représentées par des cercles rouges. AKT2 : PKB (protéine kinase B) bêta. |
Des souris protégées contre le diabète et l’obésité induits par un régime riche en lipides et en fructose
Les souris sauvages et TG ont été soumises à un régime riche en lipides et en fructose. Contrairement aux souris sauvages, les souris TG ne deviennent ni obèses ni diabétiques. Elles n’accumulent pas de TA viscéral et ne développent pas d’inflammation de ce tissu. Au niveau hépatique, la quantité de triglycérides mesurée est plus faible chez ces souris TG, qui ne développent pas de stéatose. Les marqueurs d’inflammation, de fibrose et de cytolyse hépatiques, augmentés chez les souris sauvages, restent inchangés chez les souris TG.
Conclusion
L’accumulation de lipides dans le foie provient d’une augmentation de l’entrée des acides gras dans les hépatocytes, d’une augmentation de leur synthèse et/ou d’une diminution de leur oxydation. Nous avons examiné l’effet de l’expression de différentes enzymes impliquées dans ces voies. Nos résultats [5] ont montré que PiT1 protégeait de l’accumulation de triglycérides dans les hépatocytes via une diminution de l’entrée des acides gras et une diminution de l’expression des enzymes lipogéniques, en particulier du facteur de transcription SREBP1c (sterol regulatory element binding protein 1c).
Le transporteur PiT1 serait ainsi une cible thérapeutique potentielle dans un contexte de syndrome métabolique, de diabète et d’obésité. Des études portant sur le mécanisme par lequel PiT1 module l’activité de ses partenaires pourraient ainsi permettre le développement de modulateurs efficaces.
Liens d’intérêt
L’auteur déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Le système de recombinaison Cre-lox utilise l’enzyme recombinase Cre, une tyrosine recombinase issue du bactériophage P1, afin de cibler des séquences loxP (également issues du bactériophage P1), permettant ainsi d’activer, réprimer, voire même échanger, les gènes situés entre les séquences lox.
Méthode permettant de mettre en évidence des associations de protéines après transfection des ADN complémentaires correspondants dans la levure Saccharomyces cerevisiae.
Références
- Junquero D, Rival Y. Syndrome métabolique : quelle définition pour quel(s) traitement(s) ? Med Sci (Paris) 2005 ; 21 : 1045–1053. [EDP Sciences] [PubMed] [Google Scholar]
- Perry RJ, Samuel VT, Petersen KF, et al. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014 ; 510 : 84–91. [CrossRef] [PubMed] [Google Scholar]
- Forand A, Beck L, Leroy C, et al. EKLF-driven PIT1 expression is critical for mouse erythroid maturation in vivo and in vitro. Blood 2013 ; 121 : 666–678. [Google Scholar]
- Beck L, Leroy C, Beck-Cormier S, et al. The phosphate transporter PiT1 (Slc20a1) revealed as a new essential gene for mouse liver development. PloS One 2010 ; 5 : e9148. [PubMed] [Google Scholar]
- Forand A, Koumakis E, Rousseau A, et al. Disruption of the phosphate transporter Pit1 in hepatocytes improves glucose metabolism and insulin signaling by modulating the USP7/IRS1 interaction. Cell Rep 2016 ; 16 : 2736–2748. [CrossRef] [PubMed] [Google Scholar]
- Postic C, Magnuson MA. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 2000 ; 26 : 149–150. [CrossRef] [PubMed] [Google Scholar]
- Capeau J. Voies de signalisation de l’insuline : mécanismes affectés dans l’insulino-résistance. Med Sci (Paris) 2003 ; 19 : 834–839. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Saltiel AR, Pessin JE. Insulin signaling pathways in time and space. Trends Cell Biol 2002 ; 12 : 65–71. [Google Scholar]
- Ricort JM, Tanti JF, Van Obberghen E, et al. Alterations in insulin signalling pathway induced by prolonged insulin treatment of 3T3-L1 adipocytes. Diabetologia 1995 ; 38 : 1148–1156. [PubMed] [Google Scholar]
- Xu X, Keshwani M, Meyer K, et al. Identification of the degradation determinants of insulin receptor substrate 1 for signaling cullin-RING E3 ubiquitin ligase 7-mediated ubiquitination. J Biol Chem 2012 ; 287 : 40758–40766. [PubMed] [Google Scholar]
- Yoshihara H, Fukushima T, Hakuno F, et al. Insulin/insulin-like growth factor (IGF) stimulation abrogates an association between a deubiquitinating enzyme USP7 and insulin receptor substrates (IRSs) followed by proteasomal degradation of IRSs. Biochem Biophys Res Commun 2012 ; 423 : 122–127. [PubMed] [Google Scholar]
- Shi Y, Wang J, Chandarlapaty S, et al. PTEN is a protein tyrosine phosphatase for IRS1. Nat Struct Mol Biol 2014 ; 21 : 522–527. [CrossRef] [PubMed] [Google Scholar]
- Song MS, Salmena L, Carracedo A, et al. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 2008 ; 455 : 813–817. [CrossRef] [PubMed] [Google Scholar]
© 2017 médecine/sciences – Inserm
Liste des figures
|
Figure 1. Représentation schématique de la voie de signalisation insulinique. La liaison de l’insuline à son récepteur entraîne l’autophosphorylation de celui-ci et son activation, qui induit le recrutement de protéines d’ancrage telles que l’IRS1 (insulin receptor substrat 1). L’IRS1 va être phosphorylée à son tour, ce qui va permettre le recrutement de la PI3K (phosphatidylinositol 3-kinase). Celle-ci phosphoryle les phosphatidylinositol diphosphates (PIP2) de la membrane cellulaire en phosphatidylinositol triphosphates (PIP3), activant ainsi AKT2. Cette dernière va exercer diverses actions : stimuler la synthèse de lipides et du glycogène, inhiber la production de glucose par le foie et favoriser l’entrée de glucose dans les cellules. PTEN (phosphatase and tensin homolog) possède un rôle antagoniste à la PI3K et inhibe l’activation d’AKT. Les phosphorylations sur des tyrosines sont représentées par des cercles rouges. Foxo : forkhead box O ; GSK3 : glycogen synthase kinase 3 ; SREBP : sterol regulatory element binding protein ; AKT2 : PKB (protéine kinase B) bêta. |
| Dans le texte | |
|
Figure 2. Représentation schématique de l’effet de l’invalidation de PiT1 (inorganic phosphate transporter 1) sur la signalisation insulinique. (A) En présence de PiT1, un rétrocontrôle négatif, déclenché par un signal insulinique prolongé, va permettre l’arrêt du signal. PTEN (phosphatase and tensin homolog) déphosphoryle le phosphatidylinositol triphosphate PIP3 en phosphatidylinositol diphosphate PIP2 et, ainsi, inactive la kinase AKT2. PTEN déphosphoryle également les tyrosines de l’IRS1 (insulin receptor substrat 1), ce qui libère la PI3K (phosphatidylinositol 3-kinase). De plus, le complexe USP7 (ubiquitin-specific-processing protease 7)/IRS1 se dissocie et entraîne l’ubiquitination (Ub, ubiquitine) de l’IRS1 et sa dégradation par le protéasome. (B) En l’absence de PiT1, l’expression de PTEN et son association avec l’IRS1 diminue, n’entraînant pas l’inactivation d’AKT2. De plus, USP7 reste associée à l’IRS1, la protégeant ainsi de l’ubiquitination et empêchant donc sa dégradation. Les phosphorylations sur des sérines/thréonines sont représentées par des cercles bleus. Les phosphorylations sur des tyrosines sont représentées par des cercles rouges. AKT2 : PKB (protéine kinase B) bêta. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.