")
")
| Issue |
Med Sci (Paris)
Volume 29, Number 12, Décembre 2013
|
|
|---|---|---|
| Page(s) | 1117 - 1124 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20132912015 | |
| Published online | 20 December 2013 | |
La mort cellulaire programmée ne manque pas de vocabulaire
Programmed cell death comes in many flavors
Inserm U872, équipe 19 « mort cellulaire programmée et physiopathologie des cellules tumorales », Centre de recherche des Cordeliers, Université Pierre et Marie Curie, Université Paris-Descartes, Sorbonne-Universités, UMRS 872, 15, rue de l’École de Médecine, 75006 Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Le terme apoptose est aujourd’hui le premier mot qui vient à l’esprit d’un scientifique à qui l’on parle de suicide programmé des cellules. Pourtant, l’apoptose dite classique ne constitue pas le seul programme de mort cellulaire, et de nombreuses voies dites alternatives ou même atypiques sont désormais connues. Plus qu’alternatives, ce sont parfois des voies de signalisation préférées à l’apoptose en raison du type cellulaire et/ou du contexte environnemental dans lequel se trouve la cellule (tissu, stade de développement, etc.). Dans cette revue, nous décrivons différents types de mort cellulaire programmée dont la littérature fait état à ce jour. Entre anoïkis, pyroptosis, necroptosis ou ferroptosis, vous allez découvrir que la mort cellulaire programmée ne manque pas de vocabulaire !
Abstract
Apoptosis is nowadays what comes first to your scientist mind when someone mentions cellular suicide. However this is not the sole form of programmed cell death and many other alternative or atypical pathways have now been described. These pathways are indeed rather preferred to apoptosis in some instances based on tissue origin, cell type or development stage of the target cell. In this review, we describe many different programmed cell death subtypes according to their characteristics. Discrete, brutal, final or singular cell death pathways all participate in the elimination of unwanted, damaged or dangerous cells in organisms hence contributing to our knowledge of this particular feature of living beings: dying! Through description of anoikis, necroptosis, entosis, netosis, pyroptosis or ferroptosis, we have no choice but to realize that programmed cell death comes in many flavors.
© 2013 médecine/sciences – Inserm
Il suffit de nommer la chose pour qu’apparaisse le sens sous le signe
Léopold Sédar Senghor
Homme d’État et écrivain sénégalais
Ce grand principe a été très largement utilisé par les botanistes, et connaître l’étymologie des plantes amène bien souvent à les comprendre. Il n’est pas rare que le nom, parfois barbare pour le non-initié au latin ou au grec, renseigne sur les propriétés de la plante, ou des anecdotes qui lui sont associées. De la même manière, les biologistes ont nommé les programmes de mort cellulaire. Le terme nécrose trouve son origine dans le grec nekrosis, mortification, et est d’ailleurs utilisé pour une maladie des céréales, appelée également nielle. Il est communément employé depuis le XXe siècle pour décrire la dégénérescence des tissus en médecine. Avec l’apparition du microscope, des cellules en train de mourir sont observées, et le terme nécrose est alors utilisé jusqu’en 1971 pour tous les types de mort cellulaire. Lorsque Kerr, Wyllie et Currie observent cette année-là une mort non pathologique dans certains tissus, ils la nomment « nécrose contractée » avant de se raviser et choisir apoptose en 1972. Issue du grec apo, au loin, et ptosis, chute, cette mort fait référence à la chute des feuilles. Pour l’anecdote, la chute des feuilles à l’automne sera attribuée 20 ans plus tard à la mort programmée des cellules composant leurs tiges ! Les premiers gènes régulant l’apoptose sont ensuite identifiés dans un modèle d’étude cher aux biologistes du développement : le nématode Caenorhabditis elegans [1]. Par la suite, leurs homologues sont mis en évidence dans un autre modèle de développement, la mouche Drosophila melanogaster, puis dans les cellules de souris et d’homme, faisant ainsi de l’apoptose un phénomène conservé et important qui génère, tout à coup, un engouement pour ce nouveau domaine de recherche. Cependant, l’apoptose classique, telle qu’elle a pu être décrite ces dernières années, n’est pas l’unique programme de suicide cellulaire possible. De nombreuses voies dites alternatives ou atypiques sont aujourd’hui connues. Ces voies de signalisation peuvent même être préférées à l’apoptose à cause de l’environnement de la cellule (tissu, etc.). Elles sont aussi probablement le résultat de la redondance de ce système d’autodestruction cellulaire si nécessaire à la réparation et au maintien de l’homéostasie tissulaire, ainsi qu’à l’élimination des pathogènes et à la destruction des cellules à potentiel tumoral [2]. Dans le souci de nommer pour reconnaître et classifier, les scientifiques ont, comme à leur habitude, rivalisé d’ingéniosité pour baptiser leurs découvertes, et la liste est aujourd’hui bien longue lorsqu’il s’agit de mort cellulaire. La profusion des noms nous oblige donc à la classification de ces différents types de mort.
Nous avons choisi dans cette revue de les aborder selon le déterminisme cellulaire associé : mourir discrètement, mourir brutalement, mourir finalement et mourir singulièrement. Ces notions renvoient inévitablement au concept d’immunogénicité de la mort cellulaire et, donc, à l’impact des diverses voies de mort sur le tissu, l’organe ou même l’organisme. À l’heure où l’immunothérapie antitumorale laisse entrevoir de nouveaux moyens de traiter les cancers, il est de plus en plus urgent de caractériser les effets anti- et protumoraux associés à l’induction de la mort cellulaire par chimio- et radiothérapie. Dans cette revue, nous décrirons les différents types de mort cellulaire programmée actuellement identifiés dans la littérature, après avoir brièvement rappelé les grandes caractéristiques de l’apoptose classique. La plupart des découvreurs de ces morts cellulaires s’efforcent de s’inscrire dans la lignée de Kerr, Wyllie et Currie qui avaient, à l’époque, consulté un professeur de grec ancien, James Cormack, afin de nommer la mort cellulaire programmée apoptose. Cette revue est donc aussi l’occasion de vous initier au jargon de la mort cellulaire, qui comporte désormais des termes tels que pyroptosis, anoïkis ou necroptosis.
Mourir discrètement : apoptose et anoïkis
L’apoptose est la voie de mort par excellence qui intervient lors du développement, de la morphogenèse et de l’homéostasie tissulaire. Parce qu’elle peut concerner des millions de cellules chaque jour dans un corps humain, elle doit être majoritairement tolérogène et discrète. L’élimination des cellules apoptotiques est nécessairement rapide et efficace pour remplir ces conditions. C’est notamment pour cette raison que l’apoptose n’est que très rarement, voire pas, observée in vivo, même au sein de tissus à fort renouvellement cellulaire, tels que le thymus, la moelle osseuse ou les testicules. Ce n’est qu’en inhibant la digestion des cellules par les phagocytes que l’on peut observer la présence de nombreuses cellules apoptotiques et l’accumulation de débris, cette inhibition pouvant conduire à des phénomènes d’auto-immunité dangereux pour l’organisme [3]. Dans certains cas cependant, l’apoptose s’avère très immunogène et capable d’induire des réponses immunitaires protectrices. Cette propriété est actuellement exploitée en cancérologie dans le cadre d’immunothérapie adjuvante [33] (→).
(→) Voir m/s n° 3 mars 2007, page 257
L’apoptose peut être commandée par des signaux internes à la cellule (voie intrinsèque) ou par des signaux externes (voie extrinsèque). La première voie de signalisation repose sur la perte d’intégrité membranaire des mitochondries. Ces organites renferment de nombreuses protéines capables d’induire la mort de la cellule [4]. Les membres de la famille Bcl-2 régulent la perméabilité mitochondriale, et leur expression et leur activité sont régulées par les divers stress cellulaires, tels que les dommages à l’ADN, le stress métabolique, etc. [4, 5]. La voie extrinsèque, quant à elle, implique l’engagement de récepteurs membranaires de mort, les plus courants étant Fas/CD95, TNFa receptor 1 (TNFR1) et TRAIL receptor (TRAILR) 1-2 [6]. La liaison de ces récepteurs à leurs ligands déclenche la signalisation apoptotique. Les deux voies intrinsèque et extrinsèque mènent généralement à l’activation d’une cascade d’enzymes protéolytiques, les caspases, notamment celles responsables de la morphologie cellulaire typique de l’apoptose (Figure 1). Cette cascade aboutit à la dégradation par clivage d’un millier de substrats différents, allant des protéines à activité kinase aux enzymes de la réparation de l’ADN en passant par des protéines de la réplication ou de la traduction, sans oublier des protéines de structure [7]. L’aspect de la cellule est ainsi modifié : le volume cellulaire est réduit (pyknosis), la membrane plasmique bourgeonne, la chromatine se condense, l’ADN est fragmenté (karyorrhexis) et des corps apoptotiques se forment [8]. Ces derniers sont ensuite identifiés et éliminés in vivo par des phagocytes, en partie grâce à l’externalisation des résidus phosphatidylsérine de la couche lipidique interne à la couche lipidique externe de la membrane plasmique. Chacune de ces caractéristiques constitue un outil pour détecter l’apoptose. La détection de l’apoptose par une simple observation microscopique est révolue, et les techniques actuelles sont diverses et variées. Cependant, le domaine de la mort cellulaire n’en est pas devenu plus simple, bien au contraire. Grâce en partie à l’utilisation d’inhibiteurs de caspases, des voies de signalisation sous-jacentes multiples pouvant conduire à d’autres types de mort cellulaire programmée ont été identifiées.
 |
Figure 1. Voies de signalisation conduisant à l’apoptose. L’apoptose peut être déclenchée par deux voies différentes : par la surface des cellules via les récepteurs de mort (voie extrinsèque) ou par la mitochondrie au travers de signaux internes (voie intrinsèque). Dans les deux situations, l’induction de l’apoptose passe par des caspases initiatrices, la caspase-8 ou -10 pour la voie extrinsèque, la caspase-9 formant l’apoptosome avec APAF-1 et le cytochrome c pour la voie intrinsèque. Ces caspases initiatrices activent les caspases exécutrices -3, -6 et -7 qui vont cliver des milliers de substrats différents. Cela conduit à la fragmentation de l’ADN, au bourgeonnement de la membrane plasmique et à la condensation de la chromatine. Il existe des points de convergence entre les deux voies, comme la protéine Bid, membre de la famille Bcl-2, qui active la voie mitochondriale après clivage par la caspase-8. Ce mécanisme peut être un moyen d’amplifier le signal apoptotique. Voir Glossaire pour la définition des abréviations. |
Parmi les cas particuliers d’apoptose, nous pouvons citer l’anoïkis qui ne concerne que les cellules adhérentes, car c’est une apoptose induite par le détachement cellulaire de la matrice extracellulaire. Elle implique une perte de l’attachement des intégrines β1, la diminution de l’expression de l’EGFR, l’inhibition de la signalisation ERK1 et la surexpression de BIm, un membre de la famille Bcl-2 [9, 10]. L’étude moléculaire de l’anoïkis présente un intérêt en raison de la propriété des cellules cancéreuses épithéliales invasives et métastatiques à résister à ce type de mort.
Mourir brutalement : nécrose régulée et necroptosis
Longtemps opposée à l’apoptose, la nécrose a récemment vu son statut de mort incontrôlée reconsidéré. En effet, plusieurs études ont pu démontrer l’existence de voies biochimiques complexes régulant l’exécution d’une nécrose programmée en réponse à toute une variété de signaux : les dommages à l’ADN, les excitotoxines, les ligands des récepteurs de mort, etc. La nécrose n’en reste pas moins aussi une voie passive de mort cellulaire, comme lors d’une perte d’énergie considérable ou lors de dommages drastiques irréparables. Elle se caractérise morphologiquement par un gonflement des organites intracellulaires et une rupture de l’intégrité membranaire. La nécrose est principalement pro-inflammatoire dans le tissu, ce qui pourrait constituer, dans certains cas, un avantage [11]. En effet, la nécrose s’accompagne de la libération de nombreuses cytokines et de médiateurs de l’inflammation : IL8, IL10, TNFα et HMGB1. La nécrose peut néanmoins demeurer moins immunogène que les cellules apoptotiques dites pro-inflammatoires. Elle est dans tous les cas indépendante des caspases, contrairement à l’apoptose qui peut être liée ou non à l’activation de ces protéases. Les raisons de l’absence d’activation des caspases peuvent être directes (mutation, inhibition ou suppression d’un des composants de la voie) ou indirectes (manque d’énergie pour assurer l’activation d’une voie dépendante des caspases).
Une des formes connues et bien décrites de nécrose programmée est la nécroptose [12]. Cette voie est enclenchée par l’activation des récepteurs de mort (cités ci-dessus) et des récepteurs TLR3 et TLR4. L’activation des kinases RIP1 et RIP3 est primordiale dans l’initiation de cette voie et peut être inhibée par les caspases, notamment la caspase-8, la machinerie de dégradation par ubiquitination de RIP1 et RIP3, ou par les agents pharmacologiques de type nécrostatines (Figure 2). Il est important de noter que RIP3 peut induire la mort en l’absence de RIP1, comme lors d’infections par le CMV (cytomégalovirus) par exemple. Les substrats de RIP1 et RIP3 conduisant à la mort cellulaire sont encore mal connus, et les candidats évoqués n’ont généralement pas convaincu car leur suppression n’empêche que partiellement la nécroptose. Parmi les possibles voies en aval, on trouve, entre autres, la génération d’espèces réactives de l’oxygène (ROS) par l’activation de la NADPH-oxydase 1 (NOX1), l’activation d’enzymes métaboliques de la glycogénolyse et de la glutaminolyse, et l’inhibition de la translocase mitochondriale de nucléotide adénine (ANT) [11]. L’inhibition partielle de la nécroptose lorsque l’une ou l’autre de ces voies est touchée pourrait résider dans le fait qu’elles participent toutes en parallèle à la réalisation de ce programme de mort. D’autres signaux indépendants des récepteurs de mort peuvent conduire à la nécrose régulée, notamment par l’intermédiaire de l’activation de l’enzyme de réparation de l’ADN PARP-1 [13]. Des stress génotoxiques importants, pouvant découler d’un excès de ROS, comme dans le syndrome d’ischémie-reperfusion par exemple, en sont généralement responsables [14]. L’hyperactivation de PARP-1 a de multiples conséquences, telles que l’épuisement des stocks d’ATP intracellulaire, l’activation des protéases dépendantes du calcium (calpaïnes), la libération de polymères de PAR et l’induction de la perméabilité mitochondriale [15]. Ces étapes précédentes contribuent à la sortie de l’effecteur mitochondrial de mort cellulaire AIF, qui, une fois relocalisé dans le noyau, fragmente l’ADN [16]. Cette voie de nécrose dépendante de PARP-1 a également été démontrée comme dépendante des kinases RIP1 et RIP3, soulevant la question d’une nécroptose intrinsèque, en opposition à la nécroptose extrinsèque induite par les récepteurs de mort. Certains travaux ont montré l’importance de la liaison des polymères de PAR pour la libération d’AIF sans activation des calpaïnes, se plaçant dans un type de mort baptisé parthanatos [17]. L’importance de la nécroptose a pu être démontrée dans de nombreuses conditions physiopathologiques, telles que l’inflammation de l’intestin, la perte des photorécepteurs, la pancréatite aiguë, et les infections virales [12]. Par ailleurs, son activation pourrait représenter une piste thérapeutique intéressante dans le domaine de la cancérologie.
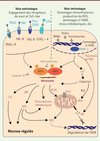 |
Figure 2. La nécrose régulée . La nécrose peut être induite par plusieurs types de signaux et comporte divers mécanismes de régulation. La nécroptose, provoquée par les récepteurs de mort et les récepteurs Toll-like, repose sur l’assemblage fonctionnel des kinases RIP1 et RIP3, à savoir la formation du nécrosome. Ce complexe est inhibé par la caspase-8 et par les molécules chimiques de type nécrostatines. La nécroptose peut impliquer des perturbations métaboliques, l’activation des kinases JNK, l’élévation du calcium intracellulaire et des espèces réactives de l’oxygène, ainsi que la perturbation de la mitochondrie, via notamment MLKL et DRP1. La nécrose peut être aussi induite par des signaux intrinsèques, tels que le stress oxydatif, l’ischémie-reperfusion, les dommages à l’ADN, tous ces événements conduisant à l’activation de PARP-1. Parmi les conséquences de l’hyperactivation de PARP-1, on trouve l’activation des protéases calpaïnes, l’activation de Bid et de Bax, la chute du potentiel mitochondrial et la libération de l’effecteur mitochondrial AIF. Cette voie intrinsèque peut aussi entraîner la formation et l’activation du nécrosome, rejoignant alors la voie extrinsèque de nécrose régulée. MNNG : N-méthyl-N’-nitro-N-nitrosoguanidine (un carcinogène). |
Mourir finalement : autophagie, catastrophe mitotique et entosis
Certains mécanismes s’apparentent en premier lieu davantage à un système de survie et de protection cellulaire qu’à un type de mort, mais ils peuvent néanmoins se solder par une mort programmée. C’est le cas de l’autophagie, la catastrophe mitotique et l’entosis. Leur immunogénicité est relativement peu caractérisée ; elle dépend probablement du type de mort impliqué. Par exemple, l’autophagie a été décrite comme nécessaire à la fois à la tolérogénicité de l’apoptose en favorisant l’exposition de signaux « eat me », et à l’induction d’une réponse immune via la libération d’HGMB1 [18].
Autophagie
Ce processus correspond à un phénomène intracellulaire d’autodigestion lysosomale des composants cytoplasmiques (organites ou cytosol), induit par des conditions de stress, telles que le manque de nutriments. L’autophagie permet à la cellule de survivre durant une période critique en recyclant les acides aminés et les acides gras de ses propres composants pour pallier les besoins énergétiques. C’est aussi un mécanisme d’élimination de certains organites, tels que les mitochondries. Il existe plusieurs sous-types d’autophagie, mais seule la macro-autophagie, caractérisée par la formation d’autophagosomes à double membrane, pourrait mener à la mort cellulaire [19] (Figure 3). Dans ce type de mort, la chromatine n’est pas condensée, le cytoplasme présente une vacuolisation très importante et les cellules ne sont pas ou peu reconnues par des phagocytes. Pour détecter l’autophagie, différentes techniques sont utilisées comme la visualisation des autophagosomes par microscopie électronique, la redistribution de LC3 au niveau des membranes d’autophagosomes ou encore l’apparition de sa forme dérivée (LC3-II) qui se distingue de la forme non modifiée (LC3-I) par une analyse sur gel de polyacrylamide [20]. L’invalidation génétique de plusieurs gènes de la famille Atg a démontré leur implication dans la régulation de cette voie. Cependant, dans les cas de carence en nutriments, leur suppression n’induit pas une diminution de la mort cellulaire, révélant ainsi le rôle primordial de l’autophagie dans la survie cellulaire. L’invalidation d’Atg5, de bécline-1/Atg6 ou d’Atg7 peut, en revanche, diminuer la mort cellulaire induite par certains agents comme l’H202 [21]. Il reste encore à clarifier le rôle de l’autophagie dans la mort cellulaire programmée, notamment sa capacité à basculer d’un mécanisme de survie à un mécanisme de mort.
 |
Figure 3. L’autophagie. L’autophagie peut être induite par diverses conditions de stress cellulaires ou lors du développement. Elle débute par la formation d’un phagophore caractérisé par une double membrane. Cette étape est finement régulée par plusieurs complexes moléculaires, comme le complexe bécline-1 et LC3/Atg8. L’élongation de la membrane autophagique s’effectue grâce au complexe Atg5/Atg12. La fusion des membranes conduit à la séquestration des organites et protéines cytoplasmiques dans une vacuole à double membrane, l’autophagosome. Les autophagosomes fusionnent ensuite avec des lysosomes pour former les autophagolysosomes, responsables de la dégradation du contenu. L’autophagie peut à la fois constituer un mécanisme de survie (manque de nutriments) ou de mort cellulaire. |
Catastrophe mitotique
Comme son nom l’indique, la catastrophe mitotique est liée à un mauvais déroulement de la mitose. Elle est observable lors de dysfonctionnements de la machinerie mitotique conduisant à une ségrégation incorrecte des chromosomes. La cellule arrête alors son cycle cellulaire et induit sa propre mort par nécrose ou apoptose, ou sa sénescence. Certaines cellules peuvent échapper à cette mort cellulaire, notamment en cas de mutations de la protéine p53, et continuent de proliférer avec les modifications chromosomiques acquises, soit par micronucléation (chromosomes ou fragments de chromosomes surnuméraires), soit par multinucléation (noyaux entiers supplémentaires) [22]. La catastrophe mitotique représente davantage un mécanisme oncosuppressif qui prévient l’apparition de cellules au contenu génomique aberrant, plutôt qu’une véritable mort cellulaire [23]. Son étude est toutefois primordiale, car plusieurs agents pharmacologiques utilisés en chimiothérapie agissent justement sur la mitose : c’est le cas du paclitaxel (Taxol®) et de la vinblastine (Velbé®) qui empêchent, respectivement, la dépolymérisation et la polymérisation des microtubules
Entosis
L’entosis (ou cannibalisme cellulaire) est observée fréquemment dans les biopsies de tumeurs, mais aussi dans d’autres conditions. Elle correspond à l’internalisation d’une cellule par une autre cellule du même type et non phagocytaire. Ce programme serait initié par une perte d’interaction avec la matrice extracellulaire [24]. Il requiert l’activation des protéines Rho et ROCK1 et n’est pas perturbé par l’inhibition des caspases ou la surexpression de la protéine anti-apoptotique Bcl-2. La cellule ingérée étant dégradée par les hydrolases lysosomales, ce programme peut être bloqué par des inhibiteurs de protéases lysosomales. Parfois, la cellule ingérée continue à se diviser et peut échapper à la mort cellulaire [25]. Seule une entosis aboutissant à la disparition définitive de la cellule peut être considérée comme une mort cellulaire programmée.
Mourir singulièrement : netosis, cornification et pyroptosis
Plusieurs types de mort cellulaire programmée décrits sont en fait des cas particuliers restreints à un type cellulaire. Ils représentent cependant des modèles d’étude intéressants, pouvant permettre d’identifier des protéines ou des mécanismes cellulaires non définis.
Netosis
La netosis ne concerne que les granulocytes neutrophiles et éosinophiles. Elle a été nommée ainsi car elle a été initiallement observée dans des cellules sécrétant des pièges extracellulaires neutrophiles (NET), constitués de protéines antimicrobiennes, de chromatine nucléaire et d’histones [26]. Cette sécrétion se produit physiologiquement en présence de micro-organismes, mais aussi de cytokines pro-inflammatoires (TNFα, IL8), de plaquettes ou encore d’auto-anticorps. Elle peut conduire à la mort des granulocytes comme on l’observe in vitro en présence de PMA. Les cellules présentent alors une importante vacuolisation cytoplasmique, une décondensation de la chromatine et une rupture de toutes les membranes nucléaires et granulaires. Cette mort ne peut pas être enrayée par les inhibiteurs de caspases ni par la nécrostatine-1, mais elle est sensible à l’inhibition de la NADPH oxydase ou de l’autophagie. La citrullination des histones pourrait participer à la netosis par l’intermédiaire de la formation des NET et de la décondensation de la chromatine, comme a pu le démontrer l’invalidation génique de PAD4 [27]. La netosis partage des caractéristiques avec la nécrose et l’autophagie, et elle pourrait être rattachée à l’un ou l’autre de ces types de mort. Comme source potentielle d’auto-antigènes, elle pourrait participer à l’installation de maladies auto-immunes.
Cornification
La cornification, qui touche uniquement les kératinocytes, est un processus de mort cellulaire physiologique requis pour la formation de la couche cornée de l’épiderme [28]. Composée de kératinocytes morts ainsi que de protéines et de lipides spécifiques, cette couche cornée confère à la peau ses fonctions de barrière. Cette mort programmée des kératinocytes, apparentée à un processus de différenciation terminale, est associée à l’activation de la caspase-14 et des transglutaminases 1, 3 et 5 [29].
Pyroptosis
La pyroptose est restreinte aux macrophages infectés par des pathogènes bactériens. Elle a pu être observée en présence de plusieurs pathogènes comme Salmonella typhimurium, Listeria monocytogenes ou Bacillus antracis. La pyroptose nécessite l’activation de la caspase-1 par une plateforme multiprotéique composée, soit de récepteurs NOD-like (NLR) ou de la protéine censeur de l’ADN cytosolique AIM2 associée à la protéine adaptatrice ASC, soit de dimères d’ASC correspondant au pyroptosome. Une fois activée, la caspase-1 induit la maturation et la sécrétion des interleukines pyrogènes IL1β et IL18, et peut parfois conduire à l’activation de la caspase exécutrice -7. Les mécanismes qui relient cette signalisation à la mort de la cellule demeurent encore non élucidés. Les caractéristiques morphologiques de la pyroptosis peuvent rappeler à la fois l’apoptose et la nécrose, et il a été suggéré aussi que la pyroptose soit plus un cas particulier d’apoptose intrinsèque qu’un type de mort à part entière.
Conclusion
Que ce soit pour mourir discrètement, brutalement, finalement ou singulièrement, toutes ces voies de mort participent à la mort cellulaire programmée dans les organismes (Tableau I). Ainsi, elles enrichissent nos connaissances sur les innombrables modalités de cette particularité du vivant : celle de mourir. La liste des voies de mort présentée dans cette revue n’est pas exhaustive, et de nouveaux types de mort continuent d’être découverts chaque année. Le dernier, en 2012, est la ferroptosis, une mort cellulaire dépendante du fer. En traitant par un nouveau composé des cellules tumorales porteuses d’une mutation de la voie oncogénique Ras, les auteurs ont induit la production d’espèces réactives de l’oxygène, la peroxydation des lipides, l’apparition de mitochondries rétrécies, puis la mort cellulaire [30]. Cette mort ne partage aucune caractéristique des autres types de mort référencés (activation des caspases, activation de Bax/Bak, augmentation du calcium intracellulaire, etc.). Il est légitime de s’inquiéter de la « surenchère néologique » de notre domaine de recherche, et il convient de modérer notre enthousiasme lors de la description de « nouvelles » voies de mort. Néanmoins, si des voies moléculaires inédites sont révélées, il faut pouvoir les nommer. Elles n’auront in fine un intérêt que si leur pertinence biologique (normale ou pathologique) est avérée. L’extraordinaire redondance des systèmes de mort cellulaire programmée que possède un organisme nous confirme le caractère essentiel de ce mécanisme pour l’intégrité du tissu, mais aussi de l’organisme tout entier [31]. Les bénéfices de la mort cellulaire programmée ne s’arrêtent d’ailleurs pas à la simple élimination de cellules dangereuses ou inutiles. Il a par exemple été montré que, chez les arthropodes et l’hydre, les cellules mourantes stimulaient la prolifération des cellules voisines par l’activité des caspases sur les voies TGFβ et Wnt, mécanisme qui participerait à la différenciation des cellules souches [32].
Classification et caractéristiques des différents types de mort cellulaire programmée présentés dans cette revue.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Glossaire
AIF : apoptosis-inducing factor
AIM2 : absent in melanoma 2
APAF-1 : apoptotic peptide activating factor-1
ASC : apoptosis-associated speck-like protein containing a CARD
Atg : autophagy related gene
Bax : Bcl-2 associated X protein
Bak : Bcl-2 homologous antagonist killer
Bcl-2 : B-cell leukemia protein-2
BIM : Bcl-2-interacting mediator of cell death
CD : cluster of differentiation
Ced : cell death abnormal
CMV : cytomégalovirus
DRP1 : dynamin related protein 1
EGFR : epidermal growth factor receptor
Egl-1 : egg laying abnormal 1
ERK1 : extracellular-regulated kinase 1
FADD : Fas associated protein with death domain
HMGB1 : high mobility group box 1
IAP : inhibitor of apoptosis protein
IL : interleukine
JNK : Janus kinase
LC3 : microtubule associated protein 1 light chain 3
LPS : lipopolysaccharide
MLKL : mixed lineage kinase domain-like protein
NADPH : nicotinamide adénine dinucléotide phosphate
NOD : nucleotide-binding oligomerization domain
NOX : NADPH-oxidase 1
PAD4 : peptidylarginine deiminase 4
PAR : poly(ADP-ribose)
PARP-1 : poly(ADP-ribose) polymerase 1
PMA : phorbol myristate acetate
p53 : protéine 53
RIP : receptor interacting protein
ROCK1 : Rho-associated kinase 1
ROS : reactive oxygen species
TGFβ : transforming growth factor-β
TLR : Toll-like receptor
TNFα : tumor necrosis factor α
TRAIL : tumor-necrosis-factor related apoptosis inducing ligand
WNT : wingless integration site
Remerciements
Les auteurs remercient vivement le Pr Hélène Merle-Béral (service d’hématologie, groupe hospitalier Pitié-Salpêtrière, Paris, France) pour sa relecture du manuscrit, et les membres de l’équipe 19 du centre de recherche des Cordeliers. Notre recherche sur la mort cellulaire programmée est financée par l’Inserm, les universités Pierre et Marie Curie et Paris-Descartes, la fondation Laurette Fugain, la fondation de France-comité cancer, l’Agence nationale de la recherche (ANR-12-EMMA-0045) et l’Institut national du cancer (INCa-5839). Lauriane Cabon est financée par des bourses de thèse de l’École normale supérieure de Cachan, la Société française d’hématologie et la fondation ARC pour la recherche sur le cancer. Ana-Carolina Martinez-Torres est financée par des bourses de thèse du CONACYT (Mexico) et la fondation ARC pour la recherche sur le cancer.
Références
- Ameisen JC. Quand la forme en une autre s’en va : la mort et la sculpture du vivant. Med Sci (Paris) 2002 ; 18 : 791–794. [CrossRef] [EDP Sciences] [Google Scholar]
- Ameisen JC. La mort cellulaire programméee : programme de mort ou programme de vie ? Med Sci (Paris) 2002 ; 18 : 109–110. [CrossRef] [EDP Sciences] [Google Scholar]
- Ravichandran KS. Beginnings of a good apoptotic meal : the find-me and eat-me signaling pathways. Immunity 2011 ; 35 : 445–455. [CrossRef] [PubMed] [Google Scholar]
- Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol 2000 ; 10 : 369–377. [CrossRef] [PubMed] [Google Scholar]
- Estaquier J, Vallette F, Vayssiere JL, Mignotte B. The mitochondrial pathways of apoptosis. Adv Exp Med Biol 2012 ; 942 : 157–183. [CrossRef] [PubMed] [Google Scholar]
- Couzinet A, Hérincs Z, Hueber AO. Régulation de la mort cellulaire programmée : vers une conception plus dynamique. Med Sci (Paris) 2002 ; 18 : 841–852. [CrossRef] [EDP Sciences] [Google Scholar]
- Meier P, Vousden KH. Lucifer’s labyrinth-ten years of path finding in cell death. Mol Cell 2007 ; 28 : 746–754. [CrossRef] [PubMed] [Google Scholar]
- Green DR. The end and after : how dying cells impact the living organism. Immunity 2011 ; 35 : 441–444. [CrossRef] [PubMed] [Google Scholar]
- Ameisen JC., Anoïkis Sci Med (Paris) 2005 ; 21 : 117. [CrossRef] [EDP Sciences] [Google Scholar]
- Taddei ML, Giannoni E, Fiaschi T, Chiarugi P. Anoikis : an emerging hallmark in health and diseases. J Pathol 2012 ; 226 : 380–393. [CrossRef] [PubMed] [Google Scholar]
- Galluzzi L, Vanden Berghe T, Vanlangenakker N, et al. Programmed necrosis from molecules to health and disease. Int Rev Cell Mol Biol 2011 ; 289 : 1–35. [CrossRef] [PubMed] [Google Scholar]
- Green DR, Oberst A, Dillon CP, et al. RIPK-dependent necrosis and its regulation by caspases : a mystery in five acts. Mol Cell 2011 ; 44 : 9–16. [CrossRef] [PubMed] [Google Scholar]
- Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose) : novel functions for an old molecule. Nat Rev Mol Cell Biol 2006 ; 7 : 517–528. [CrossRef] [PubMed] [Google Scholar]
- Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev 2006 ; 20 : 1–15. [CrossRef] [PubMed] [Google Scholar]
- Heeres JT, Hergenrother PJ. Poly(ADP-ribose) makes a date with death. Curr Opin Chem Biol 2007 ; 11 : 644–653. [CrossRef] [PubMed] [Google Scholar]
- Delavallee L, Cabon L, Galan-Malo P, et al. AIF-mediated caspase-independent necroptosis : a new chance for targeted therapeutics. IUBMB Life 2011 ; 63 : 221–232. [CrossRef] [PubMed] [Google Scholar]
- Wang Y, Kim NS, Haince JF, et al. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci Signal 2011 ; 4 : ra20. [PubMed] [Google Scholar]
- Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol 2009 ; 9 : 353–363. [CrossRef] [PubMed] [Google Scholar]
- Codogno P. L’autophagie, dégrader plus pour vivre plus, mais attention ! Med Sci (Paris) 2009 ; 25 : 323–324. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Rabinowitz JD, White E. Autophagy and metabolism. Science 2010 ; 330 : 1344–1348. [CrossRef] [PubMed] [Google Scholar]
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013 ; 368 : 651–662. [CrossRef] [PubMed] [Google Scholar]
- Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat 2001 ; 4 : 303–313. [CrossRef] [PubMed] [Google Scholar]
- Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy : mitotic catastrophe. Cell Death Differ 2008 ; 15 : 1153–1162. [CrossRef] [PubMed] [Google Scholar]
- Mailleux AA, Overholtzer M, Brugge JS. L’entose, mort cellulaire par cannibalisme entre cellules tumorales. Med Sci (Paris) 2008 ; 24 : 246–248. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- White E. Entosis : it’s a cell-eat-cell world. Cell 2007 ; 131 : 840–842. [CrossRef] [PubMed] [Google Scholar]
- Remijsen Q, Kuijpers TW, Wirawan E, et al. Dying for a cause : NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ 2011 ; 18 : 581–588. [CrossRef] [PubMed] [Google Scholar]
- Kaplan MJ, Radic M. Neutrophil extracellular traps : double-edged swords of innate immunity. J Immunol 2012 ; 189 : 2689–2695. [CrossRef] [PubMed] [Google Scholar]
- Lippens S, Denecker G, Ovaere P, et al. Death penalty for keratinocytes : apoptosis versus cornification. Cell Death Differ 2005 ; 12 : 1497–1508. [CrossRef] [PubMed] [Google Scholar]
- Denecker G, Ovaere P, Vandenabeele P, Declercq W. Caspase-14 reveals its secrets. J Cell Biol 2008 ; 180 : 451–458. [CrossRef] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis : an iron-dependent form of nonapoptotic cell death. Cell 2012 ; 149 : 1060–1072. [CrossRef] [PubMed] [Google Scholar]
- Meier P, Finch A, Evan G. Apoptosis in development. Nature 2000 ; 407 : 796–801. [CrossRef] [PubMed] [Google Scholar]
- Bergmann A, Steller H. Apoptosis, stem cells, and tissue regeneration. Sci Signal 2010 ; 3 : re8. [CrossRef] [PubMed] [Google Scholar]
- Apetoh L, Ghiringhelli F, Zitvogel L. La calréticuline détermine l’immunogénicité de la chimiothérapie et de la radiothérapie antitumorales. Med Sci (Paris) 2007 ; 23 : 257–258. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des tableaux
Classification et caractéristiques des différents types de mort cellulaire programmée présentés dans cette revue.
Liste des figures
|
Figure 1. Voies de signalisation conduisant à l’apoptose. L’apoptose peut être déclenchée par deux voies différentes : par la surface des cellules via les récepteurs de mort (voie extrinsèque) ou par la mitochondrie au travers de signaux internes (voie intrinsèque). Dans les deux situations, l’induction de l’apoptose passe par des caspases initiatrices, la caspase-8 ou -10 pour la voie extrinsèque, la caspase-9 formant l’apoptosome avec APAF-1 et le cytochrome c pour la voie intrinsèque. Ces caspases initiatrices activent les caspases exécutrices -3, -6 et -7 qui vont cliver des milliers de substrats différents. Cela conduit à la fragmentation de l’ADN, au bourgeonnement de la membrane plasmique et à la condensation de la chromatine. Il existe des points de convergence entre les deux voies, comme la protéine Bid, membre de la famille Bcl-2, qui active la voie mitochondriale après clivage par la caspase-8. Ce mécanisme peut être un moyen d’amplifier le signal apoptotique. Voir Glossaire pour la définition des abréviations. |
| Dans le texte | |
|
Figure 2. La nécrose régulée . La nécrose peut être induite par plusieurs types de signaux et comporte divers mécanismes de régulation. La nécroptose, provoquée par les récepteurs de mort et les récepteurs Toll-like, repose sur l’assemblage fonctionnel des kinases RIP1 et RIP3, à savoir la formation du nécrosome. Ce complexe est inhibé par la caspase-8 et par les molécules chimiques de type nécrostatines. La nécroptose peut impliquer des perturbations métaboliques, l’activation des kinases JNK, l’élévation du calcium intracellulaire et des espèces réactives de l’oxygène, ainsi que la perturbation de la mitochondrie, via notamment MLKL et DRP1. La nécrose peut être aussi induite par des signaux intrinsèques, tels que le stress oxydatif, l’ischémie-reperfusion, les dommages à l’ADN, tous ces événements conduisant à l’activation de PARP-1. Parmi les conséquences de l’hyperactivation de PARP-1, on trouve l’activation des protéases calpaïnes, l’activation de Bid et de Bax, la chute du potentiel mitochondrial et la libération de l’effecteur mitochondrial AIF. Cette voie intrinsèque peut aussi entraîner la formation et l’activation du nécrosome, rejoignant alors la voie extrinsèque de nécrose régulée. MNNG : N-méthyl-N’-nitro-N-nitrosoguanidine (un carcinogène). |
| Dans le texte | |
|
Figure 3. L’autophagie. L’autophagie peut être induite par diverses conditions de stress cellulaires ou lors du développement. Elle débute par la formation d’un phagophore caractérisé par une double membrane. Cette étape est finement régulée par plusieurs complexes moléculaires, comme le complexe bécline-1 et LC3/Atg8. L’élongation de la membrane autophagique s’effectue grâce au complexe Atg5/Atg12. La fusion des membranes conduit à la séquestration des organites et protéines cytoplasmiques dans une vacuole à double membrane, l’autophagosome. Les autophagosomes fusionnent ensuite avec des lysosomes pour former les autophagolysosomes, responsables de la dégradation du contenu. L’autophagie peut à la fois constituer un mécanisme de survie (manque de nutriments) ou de mort cellulaire. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.