")
")
| Issue |
Med Sci (Paris)
Volume 27, Number 1, Janvier 2011
|
|
|---|---|---|
| Page(s) | 70 - 76 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/201127170 | |
| Published online | 10 February 2011 | |
Soigner la déficience intellectuelle : la recherche d’équilibre
Curing mental retardation: searching for balance
Université du Québec à Montréal, Département de chimie, Pharmaqam, Biomed, CP 8888, Succursale Centre-ville, Montréal (Québec), H3C 3P8 Canada
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Certaines formes de déficience intellectuelle résultent d’une réduction de la capacité des neurones, en l’occurrence ceux de l’hippocampe, à moduler l’efficacité de leur transmission synaptique en réponse à certaines stimulations. Elles proviennent donc d’une altération de la fonction de certaines protéines dans les voies de signalisation qui régulent ces mécanismes dans les synapses. Afin d’augmenter les capacités cognitives des enfants présentant des déficiences intellectuelles attribuables à ces phénomènes, des travaux de recherche très prometteurs sont actuellement en cours. Ils ont pour objectif de caractériser la machinerie de signalisation dans ces états pathologiques et de sélectionner des moyens thérapeutiques susceptibles de restaurer l’état physiologique normal.
Abstract
Mental retardation (MR) occurs in 2 to 3 % of the general population and is still not therapeutically addressed. Milder forms of MR result from deficient synaptogenesis and/or impaired synaptic plasticity during childhood. These alterations would result from disequilibrium in signalling pathways regulating the balance between long term potentiation (LTP) and long term depression (LTD) in certain neurons such as hippocampus neurons. To provide mentally retarded children with increased cognitive abilities, novel experimental approaches are currently being developed to characterize signalling status associated with MR and to identify therapeutic targets that would restore lost equilibrium. Several studies also highlighted the major role played by molecular switches like kinases, phosphatases, small G proteins and their regulators in the coordination and integration of signalling pathways associated with synaptic plasticity. These proteins may therefore constitute promising therapeutic targets for a number of cognitive deficiencies.
© 2011 médecine/sciences – Inserm / SRMS
Le siège de la cognition
La déficience intellectuelle (DI) se caractérise par un déficit cognitif associé à un quotient intellectuel inférieur à 70. En Occident, 2 à 3 % de la population souffrent de diverses formes de DI. Quand ces formes sont associées à d’autres symptômes cliniques, on parle alors de formes syndromiques de DI. Si la DI se limite à ces deux caractéristiques (déficit cognitif et QI inférieur à 70), on parle, dans ce cas, de formes non syndromiques ou non spécifiques de DI. Ces différentes formes de DI peuvent avoir des origines génétiques ou environnementales. À l’échelle macro-anatomique, une DI peut être associée à une altération de la structure du cortex cérébral et de l’hippocampe, ou à une microcéphalie [1]. En revanche, certaines formes de DI, par exemple celles qui sont associées aux syndromes de Down, du X fragile, de Rett ou encore de Rubinstein-Taybi, ainsi que certaines formes non syndromiques de DI, se caractérisent par une altération de la densité et de la morphologie des épines dendritiques (Figure 1A) [1]. Les épines dendritiques sont des bourgeonnements des dendrites formant les domaines post-synaptiques qui permettent la transduction des signaux provenant des neurones pré-synaptiques. Ces structures se caractérisent par leur nature très plastique, leur morphologie et leur nombre étant modulés par l’activité neuronale. Le fait que certaines formes de DI proviendraient d’une altération de la communication neuronale et non d’une malformation structurelle du cerveau ouvre des perspectives thérapeutiques captivantes. Il semble en effet plus aisé de moduler une activité neuronale dysfonctionnelle grâce à un traitement pharmacologique administré au cours de l’enfance d’un individu que d’envisager une réparation structurelle de certaines parties de son cerveau.
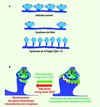 |
Figure 1 Plasticité synaptique, déficience intellectuelle et morphologie des épines dendritiques. A. Le syndrome de Rett se caractérise par une accentuation de la DLT des neurones de l’hippocampe et par une diminution de la densité des épines dendritiques. Le syndrome du X fragile se caractérise par une augmentation de la densité d’épines dendritiques longues et fines des neurones de l’hippocampe et du néocortex. B. L’établissement de la PLT est associé à une augmentation de l’efficacité de transmission synaptique des neurones de l’hippocampe, à la maturation des boutons post-synaptiques qui adoptent une morphologie en forme de champignon, à l’accumulation des récepteurs au glutamate dans les membranes post-synaptiques et à la création de nouvelles synapses (synaptogenèse). L’établissement de la DLT est associé aux processus cellulaires inverses. |
La plasticité synaptique et la cognition
Des changements morphologiques des épines dendritiques sont corrélés aux différents états d’efficacité de la transmission synaptique, ce que l’on désigne par le terme de plasticité synaptique (PS). La PS dépend de processus moléculaires permettant le maintien dans les épines dendritiques d’une optimisation de l’efficacité de transmission synaptique, que l’on appelle aussi la potentialisation à long terme (PLT), ou d’une dépréciation de l’efficacité de transmission synaptique, que l’on appelle la dépression à long terme (DLT) (Figure 1B) [2]. Les voies de signalisation qui régissent la PS mobilisent un grand nombre de récepteurs dont les récepteurs au glutamate ionotropique NMDAR (N-methyl D-aspartate receptors) et AMPAR (a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor) et les récepteurs métabotropiques mGluR. L’établissement de la PLT par les neurones de l’hippocampe répond à un stimulus conditionnant qui entraîne une augmentation de la concentration de calcium dans le bouton post-synaptique [3]. Ceci déclenche une réorganisation majeure du cytosquelette d’actine, une expansion de la tête de l’épine dendritique, un afflux de récepteurs AMPAR et des protéines qui y sont associées dans la membrane post-synaptique, une modulation de l’expression d’un certain nombre de gènes et, finalement, la formation de nouvelles épines dendritiques. Des mécanismes similaires auraient lieu lorsque le stimulus conduit à la dépression à long terme, entraînant un allongement du cou de l’épine dendritique, voire la disparition de certaines synapses (Figure 1B) [3].
Des études combinant diverses formes d’investigation (génétique, biologie moléculaire, neurophysiologie et études comportementales) appliquées à des modèles murins ont permis de mettre en évidence la relation entre les processus de plasticité synaptique, la modification morphologique des épines dendritiques et la cognition. Ainsi, les déficits cognitifs des souris chez lesquelles les fonctions des gènes NF1 (neurofibromatose de type1), α-Pix (Pak-interacting exchange factor), Ophn1 (oligophrenin 1) et Pak3 (p21-activated kinase 3)1 sont déficientes s’accompagnent d’une réduction de l’établissement de la PLT ou de la DLT (Tableau I) et d’une modification de la structure des épines dendritiques [4-7]. Les connaissances actuelles indiquent donc que les mécanismes moléculaires menant à l’établissement de la PLT et de la DLT ainsi qu’à la modification de la morphologie des épines dendritiques, sont cruciaux pour les fonctions cognitives. Ces mécanismes moléculaires des protéines de signalisation jouent le rôle de commutateurs de signalisation. Ces protéines permettent, en effet, de moduler finement la plasticité synaptique qui conduit à l’établissement de la PLT ou de la DLT et à la modification de la morphologie des épines dendritiques. Ces protéines seraient donc les garantes d’un certain équilibre de la machinerie de signalisation des épines dendritiques et des capacités cognitive de l’individu [8].
Les différentes formes de déficience intellectuelle associées à des mutations dans les régulateurs des GTPases de la superfamille des Ras.
Kinases/phosphatases et GTPases : les commutateurs de signalisation
Le rôle de commutateur de signalisation joué par des kinases et des phosphatases dans l’établissement de la PLT et de la DLT est beaucoup mieux connu que celui des GTPases [8]. L’antagonisme fonctionnel de la kinase CamKII (Ca2+/calmodulin-dependent protein kinase II) et de la phosphatase PP2B (protein phosphatase 2B) appelée aussi calcineurine en est un bon exemple [9]. Ces enzymes ont des activités antagonistes puisqu’elles phosphorylent ou déphosphorylent les mêmes cibles protéiques à la suite d’une augmentation de la concentration de calcium dans les épines dendritiques. L’affinité différente de ces protéines pour le calcium permet l’activation de la phosphatase et l’établissement de la DLT lorsque la concentration de calcium est faible, et l’activation de la kinase entraînant l’établissement de la PLT lorsque cette concentration est élevée (Figure 2A). Un grand nombre de kinases et de phosphatases interviennent dans la régulation de la PS. Les mieux étudiées sont CaMKII, PKC (protein kinase C), PKA (cAMP-dependent protein kinase), PKG (cGMP-dependent protein kinase), CKII (casein kinase II), les kinases de la cascade MAPK (mitogen-activated protein kinase), PKMζ (protein kinase M zeta), PP1 (protein phosphatase 1), PP2A (protein phosphatase 2A) et calcineurine [9]. Ces kinases et phosphatases fonctionnent en cascades et sont principalement activées en réponse aux fluctuations de la concentration des messagers secondaires : calcium, AMPc, GMPc. Elles seraient responsables du contrôle de la phosphorylation de près de 80 protéines synaptiques présentant plus de 300 sites de phosphorylation [8].
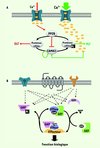 |
Figure 2 Les commutateurs moléculaires. A. Une augmentation de la concentration en calcium dans l’épine dendritique permet soit l’activation de PP2B (calcineurine), soit celle de CamKII. En effet, l’affinité de ces deux enzymes pour le complexe calmoduline/calcium étant différente, une augmentation modérée de calcium entraînerait une activation de PP2B et la déphosphorylation de ses protéines cibles, incluant CamKII (flèches rouges). Ceci participerait à l’établissement de la DLT. Inversement, une augmentation importante de la concentration de calcium (flèches vertes) entraînerait l’activation de la CamKII, son autophosphorylation rendant son activité kinase indépendante des fluctuations du calcium, le maintien des cibles sous forme phosphorylée pendant des durées prolongées et l’établissement de la PLT. B. Les GTPases alternent entre une forme inactive liée au GDP et une forme active liée au GTP. Les GAP, GDI et GEF régulent le niveau d’activation des GTPases par intégration de leurs activités antagonistes : (1) les GEF (guanine nucleotide-exchange factors) permettent l’activation de la GTPase en favorisant le remplacement du GDP par un GTP, (2) les GAP (GTPase-activating proteins) catalysent l’hydrolyse du GTP par la GTPase et permettent donc un passage rapide vers son état inactif ; et (3) les GDI (GDP dissociation inhibitors) maintiennent les GTPases dans leur état inactif et les rendent solubles en permettant la dissociation des membranes dans lesquelles elles sont ancrées. |
 |
Figure 3 La biologie des systèmes et la prédiction d’interactions génétiques. A. Les modèles de réseau d’interaction ont permis de mettre en évidence que l’étude de la fluctuation du niveau d’expression de deux gènes dans des centaines de conditions expérimentales permet de prédire avec une bonne efficacité si ces deux gènes ont un lien fonctionnel [29]. Sont représentés dans cette figure les niveaux d’expression de deux gènes dans 58 conditions expérimentales différentes (colonnes). Cette figure a été conçue avec des données réelles provenant du transcriptome du nématode Caenorhabditis elegans. Deux gènes exprimant une fluctuation transcriptionnelle similaire dans un grand nombre de conditions expérimentales différentes ont une probabilité plus élevée d’être fonctionnellement liés que deux gènes ayant une très faible corrélation de leurs profils transcriptionnels. B. De même, deux gènes codant pour des protéines ayant la capacité d’interagir ont aussi une plus forte probabilité d’être enrôlés dans les mêmes mécanismes moléculaires que deux gènes codant pour des protéines n’ayant pas cette capacité [30]. Sont représentées dans cette figure les interactions protéines-protéines identifiées dans les interactomes de Drosophila melanogaster, Caenorhabditis elegans et Homo sapiens pour 4 gènes très bien conservés entre ces organismes. Si les produits de deux gènes interagissent directement dans l’un de ces interactomes, la probabilité d’interaction fonctionnelle entre ces deux gènes est plus élevée que si aucune interaction n’a pu être détectée. Les propriétés prédictives de ces données biologiques sont cumulatives et permettent par leur association d’augmenter le niveau de certitude de la prédiction dans un réseau intégré [31]. |
Les GTPases et leurs régulateurs jouent aussi le rôle de commutateurs de signalisation. Ces protéines, regroupées en cinq familles (Rho, Rab, Arf, Ran et Ras), passent alternativement d’une forme active liée au GTP à une forme inactive liée au GDP. Elles régissent un large éventail de processus biologiques, incluant la prolifération, la différenciation et la migration cellulaires, et ce en modulant l’expression génique, la traduction, le cytosquelette et le trafic membranaire [32]. Ces protéines jouent donc un rôle crucial dans la modulation de la PS et de l’apprentissage [10]. Sous leur forme active, les GTPases traduisent les signaux cellulaires en aval d’un grand nombre de récepteurs en interagissant avec des protéines effectrices. Leur degré d’activation est assujetti à trois types de régulateurs : (i) les GEF (guanine nucleotide-exchange factors), (ii) les GAP (GTPase-activating proteins) et (iii) les GDI (GDP dissociation inhibitors). De manière analogue aux kinases et phosphatases, ces régulateurs maintiennent l’activation de leurs GTPases cibles par intégration de leurs activités antagonistes (Figure 2B). À ce jour, des mutations dans sept gènes codant pour des régulateurs des GTPases de la superfamille des Ras ont été associées à des formes syndromiques et non syndromiques de DI [10] (Tableau I). Ceci démontre le rôle essentiel joué par ces protéines dans la modulation de la PS. Ces mutations modifieraient en effet le degré d’activation et la distribution subcellulaire des GTPases, ce qui conduirait à l’altération de la synaptogenèse et de la PS dans certains neurones dont ceux de l’hippocampe [10].
La recherche d’équilibre
Une bonne compréhension des interactions synergiques ou antagonistes des différentes voies de signalisation modulant la PS et l’identification de commutateurs de signalisation comme les kinases/phosphatases, les GTPases et leurs régulateurs sont indispensables à la conception de voies thérapeutiques pour différentes formes de DI. En effet, la caractérisation du déséquilibre de signalisation associé à une mutation donnée devrait permettre d’envisager une stratégie thérapeutique susceptible d’inhiber chimiquement une protéine de signalisation ayant une activité antagoniste à celle qui est mutée. Cette hypothèse ouvre des perspectives intéressantes pour traiter les différentes formes de DI associées à des mutations dans les régulateurs des GTPases par exemple (Tableau I).
Ce principe constitue la base des approches thérapeutiques déployées afin d’améliorer les capacités cognitives des individus atteints de neurofibromatose de type I, de sclérose tubéreuse et de DI lié à l’X associé à des mutations dans le gène OPHN1. En effet, une perte de fonction de NF1 (une GAP de Ras) due à une suractivation de la voie Ras/ERK entraîne des troubles cognitifs. Ce déséquilibre renforce la signalisation inhibitrice GABAergique qui conduit à une réduction de la PLT. Deux études ont montré que les déficits cognitifs des souris Nf+/- pouvaient être significativement réduits lorsque ces dernières étaient traitées avec des agents thérapeutiques réduisant la fonction de Ras [4] ou de ERK [11]. Pareillement, la délétion chez la souris du gène Ophn1, codant pour une GAP spécifique de RhoA, conduit à une suractivation de la voie RhoA/ROCK et à une réduction de l’internalisation d’AMPAR [12]. L’altération de la PS qui en résulte est supprimée grâce à des inhibiteurs pharmacologiques de la voie RhoA/ROCK [12]. De même, les patients ayant une mutation du gène TSC2 (tuberous sclerosis complex) codant pour une GAP spécifique pour Rap1/Rheb présentent une suractivation de la voie de mTOR (target of rapamycin) conduisant à l’épilepsie, à certaines formes de DI ou d’autisme. Les troubles cognitifs associés à l’altération de TSC2 chez la souris ont été supprimés par la rapamycine, un inhibiteur de mTOR [13, 33, 34]. Ce traitement est très prometteur puisque la rapamycine est déjà utilisée en clinique contre une diversité de symptômes dont sont atteints ces patients [14]. Ces études ont clairement mis en évidence la possibilité d’abolir les déficits cognitifs, voire de rétablir les capacités cognitives en rectifiant l’équilibre au sein des voies de signalisation qui régissent les mécanismes de PS grâce à des agents pharmacologiques. Des études similaires sont actuellement en cours pour d’autres types de pathologies cognitives et/ou comportementales présentant elles aussi une altération de la plasticité synaptique : schizophrénie, autisme et maladie d’Alzheimer [15].
La mise au point d’approches thérapeutiques pour les pathologies associées à une altération de la PS en est à ses balbutiements. Afin de favoriser leur développement, une meilleure compréhension des mécanismes de signalisation et des relations fonctionnelles entre les différentes voies et protéines de signalisation est requise. La création de modèles animaux pour ces différentes pathologies associée à de nouvelles approches expérimentales de type biologie des systèmes devrait permettre de gagner ce pari (Tableau I).
La biologie des systèmes : une voie d’investigation prometteuse
Les réseaux et les systèmes de signalisation qui régissent les mécanismes cognitifs sont d’une grande complexité. Les cartographier, en caractérisant la nature des liens fonctionnels entre les différentes protéines de signalisation, est essentiel à la mise au point de nouvelles approches thérapeutiques. Les nouvelles approches issues de la génomique fonctionnelle telles que la biologie des systèmes et la biologie des réseaux ont démontré leur grand potentiel pour permettre une meilleure compréhension des mécanismes pathologiques et l’identification de nouvelles cibles thérapeutiques et d’agents pharmacologiques [16]. La biologie des systèmes regroupe des approches expérimentales qui ont pour objectif d’acquérir une connaissance globale des systèmes biologiques [35]. Ces approches supposent l’obtention de diverses informations biologiques décrivant le niveau d’expression des gènes dans le système (transcriptome), la composition protéique et métabolique du système (protéome, métabolome), la connectivité de ses différents composants (interactome). L’intégration de ces données biologiques grâce à des méthodes bio-informatiques permet de cerner la fonction de nouveaux gènes, de nouveaux modèles animaux pour des maladies humaines, des composants de voies de signalisation, des cibles thérapeutiques, des agents pharmacologiques, et de prédire les effets de ces agents [16-18]. Ces approches ont été utilisées avec succès dans différents cas de cancer et ont confirmé le rôle des kinases/phosphatases et des GTPases au sein des mécanismes d’intégration des voies de signalisation favorisant la tumorigenèse et l’acquisition de potentiel métastatique [19, 36]. Ce type d’approche commence à être appliqué aux maladies cognitives et comportementales telles que la schizophrénie, la DI et l’autisme [20, 21]. Ces études ont en particulier permis de relier la GAP SynGAP1, qui est associée à des formes non syndromiques de DI (Tableau I), à de nombreux gènes associés à la schizophrénie, suggérant que ces deux sortes de désordres cognitifs résulteraient de perturbations partageant des mécanismes moléculaires similaires [20]. Cette hypothèse a été confirmée expérimentalement, ce qui conforte la validité du recours à la biologie des systèmes pour mieux comprendre les mécanismes pathologiques associés à des désordres cognitifs et comportementaux [22]. Chez la drosophile, ce type d’étude a permis de mettre en évidence le lien fonctionnel existant entre Fmr1, un gène associé à une forme syndromique de DI, et la kinase Citron, une kinase intervenant dans le développement du cerveau chez l’homme [23]. Chez le nématode, ces études ont permis la découverte de gènes antagonistes du gène GDIa, un régulateur des GTPases Rab associé à des formes non syndromiques de DI (Tableau I) [17]. Parmi ces antagonistes, on peut citer la dystrobrévine et l’ASPM (abnormal spindle-like microcephaly-associated protein), deux protéines associées à des déficiences cognitives [17]. Bien que ces études exigent encore une validation expérimentale chez le mammifère, elles attestent tout de même de l’intérêt potentiel du recours à la biologie des systèmes pour repérer de nouvelles cibles thérapeutiques afin de traiter les désordres cognitifs et comportementaux.
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Remerciements
Nous remercions le Dr Bohrane Annabi (UQAM, département de chimie) pour la lecture critique du manuscrit. Nous remercions Julie Sylvain et Anna Y-W Lee pour les travaux graphiques des Figures 1 et 3. S. Jenna est financée par les chaires de recherche du Canada et le Conseil de recherches en sciences naturelles et en génie du Canada (CRSNG) et S. Harel par le CRSNG et le Fonds de la recherche en santé du Québec.
Ces quatre gènes sont associés à des formes de DI chez l’homme.
Références
- Dierssen M, Ramakers GJ. Dendritic pathology in mental retardation: from molecular genetics to neurobiology. Genes Brain Behav 2006 ; 5 (suppl 2) : 48-60. [CrossRef] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron 2004 ; 44 : 5-21. [CrossRef] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci 2005 ; 6 : 277-284. [Google Scholar]
- Costa RM, Federov NB, Kogan JH, et al. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 2002 ; 415 : 526-530. [CrossRef] [PubMed] [Google Scholar]
- Khelfaoui M, Denis C, van Galen E, et al. Loss of X-linked mental retardation gene oligophrenin1 in mice impairs spatial memory and leads to ventricular enlargement and dendritic spine immaturity. J Neurosci 2007 ; 27 : 9439-9450. [CrossRef] [PubMed] [Google Scholar]
- Meng J, Meng Y, Hanna A, et al. Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. J Neurosci 2005 ; 25 : 6641-6650. [CrossRef] [PubMed] [Google Scholar]
- Welzl H, D’Adamo P, Wolfer DP, Lipp HP(eds). Mouse models of hereditary mental retardation. Totowa, New Jersey USA : Humana Press, 2006. [Google Scholar]
- Lee HK. Synaptic plasticity and phosphorylation. Pharmacol Ther 2006 ; 112 : 810-832. [CrossRef] [PubMed] [Google Scholar]
- Lisman J. The CaM kinase II hypothesis for the storage of synaptic memory. Trends Neurosci 1994 ; 17 : 406-412. [CrossRef] [PubMed] [Google Scholar]
- Renieri A, Pescucci C, Longo I, et al. Non-syndromic X-linked mental retardation: from a molecular to a clinical point of view. J Cell Physiol 2005 ; 204 : 8-20. [CrossRef] [PubMed] [Google Scholar]
- Cui Y, Costa RM, Murphy GG, et al. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell 2008 ; 135 : 549-560. [CrossRef] [PubMed] [Google Scholar]
- Khelfaoui M, Pavlowsky A, Powell AD, et al. Inhibition of RhoA pathway rescues the endocytosis defects in Oligophrenin1 mouse model of mental retardation. Hum Mol Genet 2009 ; 18 : 2575-2583. [CrossRef] [PubMed] [Google Scholar]
- Jozwiak J, Jozwiak S, Grzela T, Lazarczyk M. Positive and negative regulation of TSC2 activity and its effects on downstream effectors of the mTOR pathway. Neuromolecular Med 2005 ; 7 : 287-296. [CrossRef] [PubMed] [Google Scholar]
- Franz DN, Leonard J, Tudor C, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol 2006 ; 59 : 490-498. [CrossRef] [PubMed] [Google Scholar]
- Konradi C, Heckers S. Antipsychotic drugs and neuroplasticity: insights into the treatment and neurobiology of schizophrenia. Biol Psychiatry 2001 ; 50 : 729-742. [CrossRef] [PubMed] [Google Scholar]
- Spiro Z, Kovacs IA, Csermely P. Drug-therapy networks and the prediction of novel drug targets. J Biol 2008 ; 7 : 20. [CrossRef] [PubMed] [Google Scholar]
- Lee YWA, Perreault R, Harel S, et al. Searching for signaling balance through the identification of genetic interactors of the Rab Guanine-nucleotide dissociation inhibitor gdi-1. PLOs One 2010 ; 5 : e10624. [CrossRef] [PubMed] [Google Scholar]
- McGary KL, Park TJ, Woods JO, et al. Systematic discovery of nonobvious human disease models through orthologous phenotypes. Proc Natl Acad Sci USA 2010 ; 107 : 6544-6549. [CrossRef] [Google Scholar]
- Kreeger PK, Lauffenburger DA. Cancer systems biology: a network modeling perspective. Carcinogenesis 2009 ; 31 : 2-8. [Google Scholar]
- Fernandez E, Collins MO, Uren RT, et al. Targeted tandem affinity purification of PSD-95 recovers core postsynaptic complexes and schizophrenia susceptibility proteins. Mol Syst Biol 2009 ; 5 : 269. [PubMed] [Google Scholar]
- Hsu PC, Yang UC, Shih KH, et al. A protein interaction based model for schizophrenia study. BMC Bioinformatics 2008 ; 9 (suppl 12) : S23. [CrossRef] [Google Scholar]
- Hamdan FF, Gauthier J, Spiegelman D, et al. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med 2009 ; 360 : 599-605. [CrossRef] [PubMed] [Google Scholar]
- Bauer CR, Epstein AM, Sweeney SJ, et al. Genetic, systems level analysis of Drosophila sticky/citron kinase, dFmr1 mutants reveals common regulation of genetic networks. BMC Syst Biol 2008 ; 2 : 101. [CrossRef] [PubMed] [Google Scholar]
- Bianchi V, Farisello P, Baldelli P, et al. Cognitive impairment in Gdi1-deficient mice is associated with altered synaptic vesicle pools and short-term synaptic plasticity, and can be corrected by appropriate learning training. Hum Mol Genet 2009 ; 18 : 105-117. [CrossRef] [PubMed] [Google Scholar]
- Parnas D, Haghighi AP, Fetter RD, et al. Regulation of postsynaptic structure and protein localization by the Rho-type guanine nucleotide exchange factor dPix. Neuron 2001 ; 32 : 415-424. [CrossRef] [PubMed] [Google Scholar]
- Costa RM, Yang T, Huynh DP, et al. Learning deficits, but normal development and tumor predisposition, in mice lacking exon 23a of Nf1. Nat Genet 2001 ; 27 : 399-405. [CrossRef] [PubMed] [Google Scholar]
- Guo HF, Tong J, Hannan F, et al. A neurofibromatosis-1-regulated pathway is required for learning in Drosophila. Nature 2000 ; 403 : 895-898. [CrossRef] [PubMed] [Google Scholar]
- Rennebeck G, Kleymenova EV, Anderson R, et al. Loss of function of the tuberous sclerosis 2 tumor suppressor gene results in embryonic lethality characterized by disrupted neuroepithelial growth and development. Proc Natl Acad Sci USA 1998 ; 95 : 15629-15634. [CrossRef] [Google Scholar]
- Kim SK, Lund J, Kiraly M, et al. A gene expression map for Caenorhabditis elegans. Science 2001 ; 293 : 2087-2092. [CrossRef] [PubMed] [Google Scholar]
- Cusick ME, Klitgord N, Vidal M, Hill DE. Interactome: gateway into systems biology. Hum Mol Genet 2005 ; 14 : R171-R181. [CrossRef] [PubMed] [Google Scholar]
- Zhong W, Sternberg PW. Genome-wide prediction of C. elegans genetic interactions. Science 2006 ; 311 : 14811484. [CrossRef] [PubMed] [Google Scholar]
- Ashton-Beaucage D, Therrien M. La signalisation RTK/RAS/ERK élargie : contributions de la génétique à l’assemblage d’un réseau de signalisation. Med Sci (Paris) 2010 ; sous presse [Google Scholar]
- Pallet N, Beaune P, Thervet E, Legendre C, Anglicheau D. Inhibiteurs de mTOR : des antiprolifératifs pléiotropiques. Med Sci (Paris) 2006 ; 22 : 947-952. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Le Bacquer O, Martineau Y, Mamane Y. Quand la traduction sort de sa TORpeur. Med Sci (Paris) 2006 ; 22 : 514-518. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Carvunis AR, Gomez E, Thierry-Mieg N, et al. Biologie systémique : des concepts d’hier aux découvertes de demain. Med Sci (Paris) 2009 ; 25 : 578-584. [PubMed] [Google Scholar]
- Barillot E, Calzone L, Zynovyev A. Biologie des systèmes appliqués aux cancers. Med Sci (Paris) 2009 ; 25 : 601-607. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des tableaux
Les différentes formes de déficience intellectuelle associées à des mutations dans les régulateurs des GTPases de la superfamille des Ras.
Liste des figures
|
Figure 1 Plasticité synaptique, déficience intellectuelle et morphologie des épines dendritiques. A. Le syndrome de Rett se caractérise par une accentuation de la DLT des neurones de l’hippocampe et par une diminution de la densité des épines dendritiques. Le syndrome du X fragile se caractérise par une augmentation de la densité d’épines dendritiques longues et fines des neurones de l’hippocampe et du néocortex. B. L’établissement de la PLT est associé à une augmentation de l’efficacité de transmission synaptique des neurones de l’hippocampe, à la maturation des boutons post-synaptiques qui adoptent une morphologie en forme de champignon, à l’accumulation des récepteurs au glutamate dans les membranes post-synaptiques et à la création de nouvelles synapses (synaptogenèse). L’établissement de la DLT est associé aux processus cellulaires inverses. |
| Dans le texte | |
|
Figure 2 Les commutateurs moléculaires. A. Une augmentation de la concentration en calcium dans l’épine dendritique permet soit l’activation de PP2B (calcineurine), soit celle de CamKII. En effet, l’affinité de ces deux enzymes pour le complexe calmoduline/calcium étant différente, une augmentation modérée de calcium entraînerait une activation de PP2B et la déphosphorylation de ses protéines cibles, incluant CamKII (flèches rouges). Ceci participerait à l’établissement de la DLT. Inversement, une augmentation importante de la concentration de calcium (flèches vertes) entraînerait l’activation de la CamKII, son autophosphorylation rendant son activité kinase indépendante des fluctuations du calcium, le maintien des cibles sous forme phosphorylée pendant des durées prolongées et l’établissement de la PLT. B. Les GTPases alternent entre une forme inactive liée au GDP et une forme active liée au GTP. Les GAP, GDI et GEF régulent le niveau d’activation des GTPases par intégration de leurs activités antagonistes : (1) les GEF (guanine nucleotide-exchange factors) permettent l’activation de la GTPase en favorisant le remplacement du GDP par un GTP, (2) les GAP (GTPase-activating proteins) catalysent l’hydrolyse du GTP par la GTPase et permettent donc un passage rapide vers son état inactif ; et (3) les GDI (GDP dissociation inhibitors) maintiennent les GTPases dans leur état inactif et les rendent solubles en permettant la dissociation des membranes dans lesquelles elles sont ancrées. |
| Dans le texte | |
|
Figure 3 La biologie des systèmes et la prédiction d’interactions génétiques. A. Les modèles de réseau d’interaction ont permis de mettre en évidence que l’étude de la fluctuation du niveau d’expression de deux gènes dans des centaines de conditions expérimentales permet de prédire avec une bonne efficacité si ces deux gènes ont un lien fonctionnel [29]. Sont représentés dans cette figure les niveaux d’expression de deux gènes dans 58 conditions expérimentales différentes (colonnes). Cette figure a été conçue avec des données réelles provenant du transcriptome du nématode Caenorhabditis elegans. Deux gènes exprimant une fluctuation transcriptionnelle similaire dans un grand nombre de conditions expérimentales différentes ont une probabilité plus élevée d’être fonctionnellement liés que deux gènes ayant une très faible corrélation de leurs profils transcriptionnels. B. De même, deux gènes codant pour des protéines ayant la capacité d’interagir ont aussi une plus forte probabilité d’être enrôlés dans les mêmes mécanismes moléculaires que deux gènes codant pour des protéines n’ayant pas cette capacité [30]. Sont représentées dans cette figure les interactions protéines-protéines identifiées dans les interactomes de Drosophila melanogaster, Caenorhabditis elegans et Homo sapiens pour 4 gènes très bien conservés entre ces organismes. Si les produits de deux gènes interagissent directement dans l’un de ces interactomes, la probabilité d’interaction fonctionnelle entre ces deux gènes est plus élevée que si aucune interaction n’a pu être détectée. Les propriétés prédictives de ces données biologiques sont cumulatives et permettent par leur association d’augmenter le niveau de certitude de la prédiction dans un réseau intégré [31]. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.